 Genetik, Molekularbiologie & Evolutionsfaktoren
Genetik, Molekularbiologie & Evolutionsfaktoren
24.10.23 Beobachtungen in der freien Wildbahn stellen die Rolle der Selektion in Frage
Seit Charles Darwin wird der natürlichen Selektion meist eine Hauptrolle beim evolutionären Wandel zugebilligt. Eine aktuelle Langzeitstudie an Eidechsen der Gattung Anolis brachte diesbezüglich eine Überraschung: Die Daten zeigen, dass in diesem Fall nicht die natürliche Auslese das Vorhandensein von Merkmalen steuert, und dass sie bei den untersuchten Eidechsen in der freien Wildbahn längerfristig kaum eine Rolle spielt.
Die Idee der Evolution ist sehr alt und kein neuzeitlicher Gedanke. Bereits Anaximander, ein griechischer Philosoph des 7. Jahrhunderts v. Chr., vertrat die Ansicht, dass der Mensch von Tieren abstammt, wahrscheinlich von Fischen (Encyclopedia Britannica 2023). Dies ist insofern dem heutigen Evolutionsverständnis nicht unähnlich, als hier ebenfalls die Menschen einen gemeinsamen Vorfahren mit den Fischen hatten, wenn wir weit genug in der Zeit zurückgehen. Heute versteht man unter Evolution „in erster Linie die biologische Evolution. Darunter wird die von Generation zu Generation stattfindende allmähliche Veränderung der vererbbaren Merkmale einer Population von Lebewesen und von anderen organischen Strukturen (z. B. Viren) verstanden“ (Wikipedia 2023).
Die Evolution, so glaubt man, ist ein realer biologischer Prozess, der alle Organismen hervorgebracht hat, die jemals auf der Erde gelebt haben. Allerdings ist bis heute umstritten, ob ein Mechanismus bekannt ist, mit dem man Höherentwicklung (Innovationen) erklären kann. Wenn alle höheren komplexen Lebensformen aus einfacheren entstanden sind, muss es eine treibende Kraft geben, die diesen Prozess vorantreibt. Eine Kraft, die Gottes schöpferisches Wirken ersetzen könnte. Im Jahr 1859 wurde dieses Problem angeblich von dem britischen Theologen und Naturforscher Charles Darwin gelöst, der argumentierte, dass die Evolution durch einen Prozess angetrieben wurde, den er natürliche Selektion nannte. Die natürliche Auslese als treibende Kraft der Evolution ist der Kernpunkt von Darwins Evolutionsverständnis. Und er glaubte, sie sei gleichsam so kreativ wie Gott, denn er schrieb:
„Man kann sagen, dass die natürliche Auslese täglich und stündlich in der ganzen Welt jede noch so kleine Veränderung prüft, das Schlechte verwirft, das Gute bewahrt und addiert, still und unmerklich, wann und wo immer sich die Gelegenheit bietet, an der Verbesserung jedes organischen Wesens in Bezug auf seine organischen und anorganischen Lebensbedingungen arbeitet. Wir sehen nichts von diesen langsamen, fortschreitenden Veränderungen, bis der Zeiger der Zeit den langen Ablauf der Zeitalter markiert hat, und dann ist unser Blick in längst vergangene geologische Zeitalter so unvollkommen, dass wir nur sehen, dass die Lebensformen jetzt anders sind als sie früher waren“ (Darwin 1859).
Es ist klar, dass Darwin die Evolution für einen realen Prozess hielt. Allerdings entzieht er sich weitgehend der Beobachtung, weil er so unvorstellbar langsam abläuft. Bis Wissenschaftler begannen, die Veränderungen und Adaptionen von Organismen in Langzeitstudien genauer zu untersuchen, war dies der Standard der Evolutionsbiologie. Das änderte sich in den letzten 40 Jahren, als eine explosionsartige Zunahme von Studien bewies, dass Veränderungen sehr schnell ablaufen können – sogar von einer Generation zur nächsten. Besonders gut untersucht ist dies bei Anolis-Eidechsen, die vor allem in der Karibik und angrenzenden Gebieten vorkommen. Diese können sich sehr schnell an neue Biotope anpassen, beispielsweise auf neu besiedelten Inseln. Eine neue Studie, die in der Zeitschrift PNAS vorgestellt wurde, unterstreicht diese Fähigkeit erneut.
James Stroud, Assistenzprofessor an der School of Biological Sciences des Georgia Institute of Technology (USA), hat mit einem Team in einer Langzeitstudie an einer Population von Eidechsen gemessen, wie ihre Evolution in freier Wildbahn verläuft (Stroud et al. 2023). Was er herausfand, stellt eine lang gehegte evolutionäre Überzeugung in Frage, nämlich die Rolle der natürlichen Selektion.
Stroud und sein Team führten eine Feldstudie durch, bei der vier verschiedene Arten von Anolis-Eidechsen auf einer kleinen Insel in den Fairchild Tropical Botanic Gardens in Coral Gables, Florida, beobachtet wurden. Er bestimmte das Überleben dieser vier Eidechsenarten in fünf aufeinanderfolgenden Zeiträumen. Bei den eingefangenen Exemplaren führte er weitere morphologische Analysen im Labor durch, wo er Schädel, Beine, Füße, das Gewicht und sogar die Haftfähigkeit der Zehen der Eidechsen verglich. Nachdem die Echsen mit einer Identifikationsnummer versehen und mit einem winzigen Etikett unter der Haut markiert worden waren, entließ das Team die Eidechsen auf die gleichen Äste, in denen sie sie gefunden hatten. Drei Jahre lang wurden Eidechsen gefangen, Messungen durchgeführt, die Eidechsen wieder freigelassen und untersucht, welche Eidechsen überlebten.
Nach drei Jahren konnte das Team die Überlebensdaten mit der Variation von Körpermerkmalen in Beziehung setzen und auf diese Weise analysieren, welche Körpermerkmale wichtige Voraussetzungen für das Überleben sind. Theoretisch könnten die Analysen zu einem besseren Verständnis dazu führen, wie die natürliche Auslese auf Merkmale der gesamten Gruppe der Eidechsen wirken würde. Barzler kommentiert (in Übersetzung): „Zu seiner Überraschung stellte Stroud fest, dass die stabilisierende Form der natürlichen Selektion – diejenige, die die gleichen, durchschnittlich ausgeprägten Merkmale einer Art beibehält – äußerst selten war. Tatsächlich variierte die natürliche Selektion im Laufe der Zeit massiv. In manchen Jahren überlebten Eidechsen mit längeren Beinen besser, in anderen Jahren Eidechsen mit kürzeren Beinen. Zu anderen Zeiten gab es überhaupt kein klares Muster.“
„Das faszinierendste Ergebnis ist, dass die natürliche Selektion im Laufe der Zeit extrem variabel war“, so Stroud et al. (2023). „Wir haben oft gesehen, dass die Selektion von einem Jahr zum nächsten eine völlig andere Richtung einschlug. Wenn man sie jedoch zu einem langfristigen Muster zusammenfasst, heben sich all diese Schwankungen praktisch von selbst auf: Die Arten blieben über den gesamten Zeitraum hinweg bemerkenswert ähnlich“ (nach Barzler 2023).
Die in PNAS dokumentierten Ergebnisse werden als „überraschend“ und „noch nie dagewesen“ bezeichnet. Langzeitstudien wie die von Stroud et al. wurden bisher aufgrund des hohen Arbeits- und Zeitaufwands kaum durchgeführt (Barzler 2023).
Die Daten zeigen also, dass in diesem Fall nicht die natürliche Auslese das Vorhandensein von Merkmalen steuert, und dass sie bei den untersuchten Eidechsen in der freien Wildbahn längerfristig kaum eine Rolle spielt. Man kann gespannt sein, ob weitere Langzeitstudien dieser Art zu ähnlichen Ergebnissen führen.
Die Rolle der natürlichen Selektion, die für Darwins Theorie und Nachfolgetheorien zentral war und ist, wurde von vielen Biologen zu Lebezeiten Darwins und auch danach kritisch gesehen, was wenig bekannt ist (Thomas 2021). Das betrifft vor allem das vermeintliche kreative Potenzial der Selektion. Schon vor Darwin schätzte Edward Blyth, ein britischer Biologe, bereits im Jahr 1835 die Wirkung der Selektion, die er wie Darwin „Zuchtwahl“ nannte, wie folgt ein: „Dasselbe Gesetz [der Zuchtwahl], das von der göttlichen Vorsehung festgesetzt wurde, um die typischen Merkmale einer Art zu erhalten, kann vom Menschen einfach umgedreht werden in einen Mechanismus, um verschiedene Abarten [d. h. Varianten] zu züchten. Aber es ist auch deutlich, dass, wenn der Mensch den sexuellen Verkehr bei diesen Rassen nicht regulieren würde, sie alle bald zum ursprünglichen Typ zurückkehren würden“ (Blyth 1835).
Dennoch hat sich die Sicht durchgesetzt, dass Selektion eine formende, gleichsam schöpferische Kraft sei – und das, obwohl eine Auswahl („Selektion“) zuerst das Vorhandensein von Varianten erfordert, aus denen „gewählt“ werden kann. Die neue Langzeitstudie an den Anolis-Eidechsen hat diese Sicht einmal mehr nicht bestätigt. In diesem Fall war zudem überraschend, dass nicht einmal eine bestimmte Richtung über mehrere Generationen hinweg erkennbar ist. Auch eine so umfassende, arbeitsintensive Studie über den Einfluss der Selektion bei Anolis-Echsen konnte also nicht zeigen, dass Selektion Innovationen erzeugt hätte.
Quellen
Blyth E (1835) And attempt to classify the Varieties of Animals, with observations on the marked seasonal and other changes which naturally take place in various British species, and which do not constitute varieties. Magazine of natural History, 8(1), 40-53.
Darwin C (1859) On the Origin of Species. Kapitel 4. Natural Selection. Zitat aus: https://www.theguardian.com/science/2008/feb/09/natural.selection
Encycopedia Brittanica “Anaximander” (2023) https://www.britannica.com/biography/Anaximander
Barzler C (2023) Long-term lizard study challenges the rules of evolutionary biology. October 9, 2023. https://phys.org/news/2023-10-long-term-lizard-evolutionary-biology.html
Stroud JT, Moore MP, Langerhans B & Losos JB (2023) Fluctuating selection maintains distinct species phenotypes in an ecological community in the wild. Proc. Natl. Acad. Sci. 20 (42), e2222071120 https://www.pnas.org/doi/10.1073/pnas.2222071120
Thomas N (2021) Taking leave of Darwin. A Longtime Agnostic Discovers the Case for Design. Seattle: Discovery Institute Press.
Wikipedia: „Evolution“. https://de.wikipedia.org/wiki/Evolution
Autor dieserNews: Peter Borger
© 2023, http://www.genesisnet.info/schoepfung_evolution/n323.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
12.10.23 Repetitive DNA-Sequenzen steuern die Ausprägung von Genen
Erneut wurde eine wichtige bisher unbekannte Funktion der „Junk-DNA“ aufgeklärt
Kurze Tandemwiederholungen (KTW, engl. Short Tandem Repeats) sind im gesamten Genom (Erbgut) vorhanden, entweder in codierenden Sequenzen, oder außerhalb und zwischen den Genen. Ihre Funktionen waren lange Zeit unklar, und sie wurden von Evolutionsbiologen oft als „Junk-DNA“ katalogisiert, d. h. als Sequenzen ohne biologische Funktion. Zunehmend werden sie jedoch durch Genetiker als regulatorische Elemente des Genoms erkannt. Eine neue in Science veröffentlichte Studie zeigt nun, dass 5 % des Genoms aus solchen KTW aufgebaut sind und wichtige Funktionen bei der Expression (Nutzung) von genetischer Information haben, insbesondere von proteincodierenden Genen.
Genetische Elemente des Genoms
Das Genom eines Organismus ist die Gesamtheit der in der DNA gespeicherten genetischen Information, die in einer Zelle vorhanden ist. In der Geschichte der Erforschung der DNA wurden zuerst proteincodierende Sequenzen nachgewiesen (kurz „Proteingene“), die sozusagen für die Werkzeuge der Zellen codieren und etwa 20 Prozent der gesamten DNA ausmachen (einschließlich der Introns und anderer regulatorischer Sequenzen, die für einen funktionsgerechten Aufbau der codierten Werkzeuge benötigt werden). Darüber hinaus enthält das Genom Sequenzen, die auf den ersten Blick keine Funktion zu haben scheinen, weil sich darin kurze Nukleotidabfolgen in Tausenden von Kopien – manchmal Zehntausenden – wiederholen. Diese repetitiven Elemente sind als „long interspersed nuclear elements“ (LINE; 21% des menschlichen Genoms), endogene Retroviren (ERV; 8% des Genoms) und Alu-Sequenzen bekannt. Alu-Sequenzen werden zu den „short interspersed nuclear elements“ (SINE; etwa 11% des Genoms) gerechnet. Dabei handelt es sich um kurze DNA-Abschnitte, die ihren Namen (Alu-Sequenzen) ursprünglich durch die Wirkung des Restriktionsenzyms von Arthrobacter luteus (Alu)* erhielten, das gezielt nur diese Sequenzen schneidet.* Zusammen machen diese Elemente fast die Hälfte des gesamten menschlichen Genoms aus (Borger 2018). Diese repetitiven Sequenzen wurden lange Zeit als funktionslose Überbleibsel des Evolutionsprozesses betrachtet (z. B. als Überbleibsel von uralten im Genom integrierten Viren) und als „egoistische DNA“ oder „Junk-DNA“ bezeichnet, da damals ihre einzige bekannte Funktion zunächst nur die Selbstreproduktion war. Zunehmend wurde jedoch erkannt, dass es sich um regulatorische Sequenzen für die Nutzung von Genen (Genexpression) und die Induktion von Variation handelt (Borger 2018; 2023).
Eine vierte Klasse von repetitiven genetischen Elementen sind die sog. „short tandem repeats“ (kurze Tandemwiederholungen; KTW). Auch bekannt als „Mikrosatelliten“ oder „einfache Sequenzwiederholungen“ handelt es sich dabei um DNA-Elemente, die aus einer sehr kurzen sich wiederholenden Einheit von 1 bis 6 Nukleotiden (DNA-Buchstaben) bestehen. Oft haben die KTW-Sequenzen aber auch Längen von bis zu 100 Nukleotiden und sie sind bei Prokaryoten und Eukaryoten, einschließlich des Menschen, weit verbreitet. Das menschliche Genom besteht zu etwa 5 % aus diesen KTW-Elementen. Sie sind im gesamten Genom zu finden – sogar in den codierenden Regionen von Proteingenen, wo sie für längere Aminosäureketten mit denselben oder nur zwei Aminosäuren codieren. Da DNA-Wiederholungen einen geringen Informationsgehalt haben, wurden sie von Evolutionsbiologen lange Zeit als funktionslos betrachtet. In einem kürzlich erschienenen Artikel in der Wissenschaftszeitschrift Science zeigen Molekulargenetiker nun, dass KTW als Andockstellen für Proteine dienen, die die Genexpression steuern (Horton 2023, Kuhlman 2023).
Kurze Tandemwiederholungen (KTW) als genetische Schalter
Zur Herstellung von Proteinen wird die Information eines Gens in ein Botenmolekül (mRNA) umgeschrieben. Dieser Prozess wird von Transkriptionsfaktoren (TF) durchgeführt; das sind Proteine, die an DNA-Sequenzen im Genom binden. Im Laufe der Jahre wurden Hunderttausende solcher „Andockstellen“ identifiziert. Dennoch binden viele TF an DNA-Abschnitte, denen eine solche Andockstellen fehlt, während andere DNA-Abschnitte mit solchen Andockstellen von TF unbesetzt bleiben. Dies deutet darauf hin, dass zusätzliche DNA-Sequenzen eine Rolle bei der Steigerung bzw. Regulation der Genexpression spielen. Die Autoren des Science-Artikels zeigen, dass die KTW, die oft in so genannten Enhancern (Transkriptionsverstärker) angereichert sind, diese Rolle übernehmen (Kuhlman 2023, Horton 2023).
Die Forscher untersuchten die Bindungsstärke von zwei grundlegenden TF (Pho4 und MAX), die an eine CACGTG-DNA-Sequenz („Gene-switch“) binden, und bewerteten die Auswirkungen mehrerer verschiedener Arten von benachbarten KTW auf ihre Bindungsstärke an die DNA und ihre Aktivität. Sie fanden eine bis zu 70-fach erhöhte Bindung für 609 verschiedene TF-DNA-Kombinationen in Gegenwart verschiedener KTW. Weitere Analysen ergaben, dass etwa 90 % der untersuchten TF bevorzugt mindestens einen Typ von KTW binden. Da KTW in hohem Maße veränderbar sind, schlagen sie vor, sie als eine „leicht evolvierbare Klasse“ von regulatorischen DNA-Elementen zu betrachten. Damit ist gemeint, dass durch die Veränderung der Länge der KTW-Sequenzen leicht neue Genexpressionsmuster erzeugt werden können; d. h. die Expression von Genen kann dadurch dirigiert werden. Mit anderen Worten: KTW erzeugen Variation und sollten daher als Variation-induzierende genetische Elemente bezeichnet werden (Borger 2018; 2023). Da bevorzugte KTW nicht unbedingt bekannten DNA-Bindungsmustern ähneln müssen, vermuten die Forscher, dass es einen Mechanismus geben könnte, durch den ähnliche TF in verschiedenen regulatorischen Regionen rekrutiert werden können, damit sie unterschiedliche Gene regulieren. Sie schlagen vor, dass KTW die Muster beherbergen und als zusätzliche „Gen-Schalter“ dienen, um die lokale TF-Konzentration und die Bindungsreaktionen abzustimmen, und so die Genexpression abstimmen (Horton 2023). Einmal mehr zeigt sich, dass das Genom viel komplexer ist als bisher angenommen und dass scheinbar nutzlose DNA-Sequenzen Funktionen – beispielsweise genregulatorische – besitzen.
Fazit
Mit dem heutigen Wissen muss man eine Zelle als einen Nanocomputer betrachten. Das Genom der Zelle erweist sich als ein informationstragendes und -verarbeitendes System, das eine begrenzte Anzahl von Informationssequenzen enthält, die als proteincodierende Gene (also für die zellulären Werkzeuge) codieren, sowie eine große Zahl von regulatorischen Informationssequenzen, die helfen, dass die „Werkzeuge“ in den passenden Mengen zur richtigen Zeit exprimiert werden. Einige Molekularbiologen haben das Genom als proteincodierende Inseln in einem Ozean von regulatorischen Schaltern bezeichnet. Andere beschreiben Genome als RNA-Computer mit Protein-Output. Zellen sind programmiert. In der Biologie werden zunehmend Ausdrucksweisen verwendet, die auch in der Informatik und Kybernetik zu finden sind. Dieser Sprachgebrauch verdeutlicht, dass in den Biowissenschaften zunehmend auch der Aspekt der Informationsverarbeitung Raum gewinnt.
Bauen sich Computer von alleine? Werden Computer durch zufällige (molekulare) Prozesse programmiert? Selbst die einfachste Zelle ist noch mehr als ein gigantischer Computer, der Eingangssignale aus seiner Umgebung aufnimmt und sie integriert, um eine angemessene biologische Leistung hervorzurufen. Wir wissen, dass Computer und Computerprogramme nur durch Intelligenz entworfen werden können. Dies gilt für alle Informationssysteme, auch für diejenigen, die in den Zellen vorhanden sind.
Quellen
Borger P (2018) Darwin Revisited – Or how to understand biology in the 21st century. Scholars Press. pp 142–163.
Borger P (2023) Über den Entwurf des Lebens. Mobile genetische Elemente. Studium Integrale J. 30, 22–30.
Horton CA et al. (2023) Short tandem repeats bind transcription factors to tune eukaryotic gene expression. Science 381, eadd1250. https://www.science.org/doi/full/10.1126/science.add1250?intcmp=trendmd-sci
Kuhlman TE (2023) Repetitive DNA regulates gene expression. Science 381, 1289–1290. (editorial) https://www.science.org/doi/full/10.1126/science.adk2055
Autor dieserNews: Peter Borger
© 2023, http://www.genesisnet.info/schoepfung_evolution/n322.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
25.09.23 Sprunghafte Entstehung von Genen?
Zunehmend wird beobachtet, dass unterschiedliche Organismen ihre eigenen, einzigartigen Gene haben, die so genannten de-novo-Gene. Wirklich neu sind sie aber nicht, sondern in nicht-funktionaler Form bereits latent (verborgen) vorhanden. Ihre latente Existenz in der DNA war unerwartet und widerspricht den gängigen Vorstellungen gradueller Evolution. Evolutionstheoretiker sind nun gezwungen, davon auszugehen, dass Genome Lagerstätten voller potenzieller Funktionen sind – eine Vorstellung, die eher mit einem vorausschauenden Schöpfer vereinbar ist.
Die Entstehung neuer proteincodierender Gene aus zufällig erzeugten DNA-Sequenzen ist statistisch eher unwahrscheinlich (Lau & Dill 1990). Das liegt daran, dass die Strukturform von Proteinen mit einer beliebigen Sequenz unspezifisch und ungeordnet ist; eine solche Struktur kann Amyloid-Plaques bei Alzheimer und neurodegenerativen Erkrankungen ähnlich sein. Eine zufällig erzeugte DNA-Sequenz würde also in der Regel nicht für funktionale Proteine codieren, sondern eher schädlich sein (Brown 2023). Deswegen suchten Evolutionstheoretiker die Entstehung neuartiger genetischer Information (neuer Gene) bisher in einem Prozess, der mit einer Genduplikation (Genverdopplung) startet. Anschließend muss eines der Duplikate (oder beide) durch Mutation verändert werden und sich durch Selektion in der Population durchsetzen. Die Genese von neuen Genen wäre damit nicht nur ein gradueller, sondern auch ein extrem langwieriger Prozess. Das Motto lautet: Natura non facit saltus – „Die Natur macht keine Sprünge“. Dies erklärt auch die Popularität der Darwin’schen Evolutionslehre.
In weniger als einem Jahrzehnt wurde diese Ansicht jedoch durch umfangreiche Indizien für das saltatorische (sprunghafte) Auftreten von neuen Genen in Frage gestellt (Borger 2020). Eine große Anzahl von Genen scheint ohne evolutionäre Vorgeschichte, sozusagen aus dem Nichts – de novo – entstanden zu sein. Natürlich entstehen Gene nicht wirklich aus dem Nichts. Unter einem de-novo-Gen versteht man vielmehr ein funktionales Gen, das zuvor funktionslos war. In der Evolutionsbiologie wird bereits dann von einem de-novo-Gen gesprochen, wenn es nur bei einer von zwei verwandten Arten auftritt – es wird dann postuliert, dass es evolutionär de novo entstanden ist. Das Neue ist also nicht das Gen an sich (eine DNA-Sequenz), sondern eine neu auftretende Funktion, ausgehend von einem zuvor (mutmaßlich) funktionslosen Gen.
De-novo-Gene kommen in vielen eukaryotischen Arten vor und codieren Proteine, die in vielen Geweben exprimiert werden und reich an Protein- und DNA-Bindungsdomänen sind. Anders ausgedrückt: Sie sind funktional, d. h. sie sind an den molekularbiologischen Wechselwirkungen in den Zellen beteiligt. Eine unerwartete Quelle genetischer Information liegt in den scheinbar nutzlosen Regionen, die lange Zeit als „Müllhalden“ („junk yards“) der Evolution interpretiert wurden. In einer aktuellen Arbeit in Nature Ecology & Evolution vermuten Evolutionstheoretiker, dass bei Menschen de-novo-Gene aus DNA-Abschnitten entstanden seien, die für scheinbar nutzlose RNA-Transkripte in Makaken codieren, nämlich sogenannte lange nicht-codierende RNA, lncRNA (An et al. 2023).
De-novo-Gene beim Menschen
Die Autoren beschreiben, wie sie 45 spezifisch menschliche de-novo-Gene entdeckten, also beim Menschen vorkommende funktionale Gene, für die bei Makaken keine Funktion bekannt ist. Sie identifizierten charakteristische U1-Elemente und RNA-Spleiß-verwandte Sequenzen, die für den RNA-Kernexport verantwortlich sind; in dieser Hinsicht unterscheiden sich die mRNAs von lncRNAs. Die lncRNAs haben alle ein funktionales U1-Element, das bei den mRNAs fehlt (An et al. 2023). Die Forscher wählten eines dieser Gene aus (ENSG00000205704), das in der Gehirnentwicklung exprimiert (ausgeprägt) wird. Damit wiesen sie experimentell nach, dass das Ausschalten oder die Überexpression des Gens in menschlichen embryonalen Stammzellen die neuronale Reifung von kortikalen Organoiden (d. h. ein in Kulturgefäßen gezüchtetes 3-dimensionales Gebilde aus neuronalen Zellen, die als Minimodell des Gehirns dienen) beschleunigt bzw. verzögert. Wenn das Gen in Mäuse übertragen und exprimiert wurde, entwickelten diese Gehirne mit einer höheren kortikalen Faltungsstruktur, die für die Gehirnmorphologie der menschlichen Großhirnrinde typisch ist (Brown 2023). Beim Menschen produzieren die funktionalen de-novo-Gene mRNAs, die den Zellkern aktiv verlassen, und die Proteine, für die sie codieren, werden ordnungsgemäß übersetzt und tragen direkt zur Entwicklung des menschlichen Gehirns bei (An et al. 2023). Wie die sprunghaften Umwandlungen von lncRNA-Gene in vollwertige Protein-Gene, die wir nur beim Menschen finden, zustande kommen, ist bisher völlig ungeklärt.
Die Entstehung voll funktionsfähiger Gene in einem Sprung wird als ein Alles-oder-Nichts-Typ der Emergenz verstanden, d. h. als ein plötzliches, unableitbares bzw. unvorhersehbares Ereignis, und entkräftet Theorien der allmählichen evolutionären Exaptation (Übernahme neuer Funktionen in einem neuen Zusammenhang). Aus diesem Grund wurde 2017 auch in der Genetik das Modell der Voranpassung („pre-adaptation“) aufgestellt, das die Präexistenz funktionaler Merkmale in bislang funktionslosen Genen und eine Alles-oder-Nichts-Entstehung zur Funktionalität beinhaltet (Wilson et al. 2017). Dieses Modell erklärt allerdings nichts, sondern ist nur eine alternative Beschreibung der Beobachtungen. Es bleibt die Frage: Wie kann Funktion bereits in ungenutzter „Junk-DNA“ vorhanden bzw. vorangepasst sein?
Genome werden zunehmend als große, dynamische und vorangepasste Reservoirs betrachtet, die neue Strukturen und Funktionen hervorbringen können. In diesen Reservoirs stecken also nach dieser Sicht latent neue Strukturen und Funktionen, die durch geringfügige Änderungen zur Ausprägung kommen können. Auf diese Weise soll Evolution nicht (nur) an alten Teilen „herumbasteln“, sondern nach den Vorstellungen der Evolutionsbiologen auch gleichsam neue Dinge erschaffen (Karger et al. 2018). Aber was heißt hier „neu“ genau? Schließlich ist die Bildung neuartiger funktionaler Gene aus nicht-codierender DNA aus Sicht der materialistischen (neo-)darwinistischen Sicht der Evolution, in der Zufallsmutationen und Selektion die einzigen bekannten Wirkprinzipien sind, kaum zu erwarten. Wie sollten bislang funktionslose Zufallssequenzen durch eine Anhäufung von Zufallsmutationen über Millionen von Jahren funktionsfähig werden? Ein solches Szenario wäre nicht nur aus empirischer, mathematischer und informationstechnischer Sicht, sondern auch aus evolutionärer (materialistischer) Sicht völlig unrealistisch. Die Situation stellt sich anders dar: Man kann heute davon ausgehen, dass in den Genom-Netzwerken der Lebewesen Voranpassungsmechanismen existieren. Das jedoch passt besser in den Rahmen eines intelligenten Designs. Die überraschende Entdeckung, dass neue Gene sprunghaft aus nicht-proteincodierenden Sequenzen hervorkommen können, deutet darauf hin, dass die DNA mit vorangepassten Sequenzen und nicht mit nutzlosem Müll ausgestattet sein könnte.
Deutung im Schöpfungsrahmen
Wie die neuen Entdeckungen in das Paradigma der Schöpfungswissenschaft passen, muss weiter untersucht werden. Es existiert durchaus die Möglichkeit, dass die lncRNA bei Makaken gar nicht funktionslos sind, sondern bisher unbekannte Funktionen im Zellkern haben, wie die U1-Sequenz zeigt, die sicherstellt, dass die betreffende lncRNA im Zellkern verbleibt. Im evolutionären Rahmen haben sich Makaken aus der Familie der Meerkatzenverwandten und Menschen vor grob 30 Millionen Jahren auseinanderentwickelt (vgl. Scholl 2023, Tab. 1). Funktionslose Sequenzen würden dies aufgrund der zufälligen Anhäufung von Mutationen so lange Zeit nicht unbeschadet überleben. Es ist also anzunehmen, dass die lncRNA-Gene bei Makaken funktional sind. Beim Menschen sind sehr ähnliche proteincodierende Gene für die Gehirnentwicklung ebenfalls funktional. Die evolutionäre Verwandtschaft von Menschen und Makaken wird wie üblich einfach unbewiesen vorausgesetzt, d. h. die menschlichen proteincodierenden Gene sollen in dieser Sichtweise aus lncRNA-Genen hervorgegangen sein, wie sie in Makaken vorgefunden werden. Die Beobachtungen sprechen jedoch nicht für die schrittweise Darwin’sche Evolution, wie sie immer noch in Schulen und Universitäten gelehrt wird. Die Entstehung von funktionalen Genen ausgehend von nicht-funktionalen Genen (also in diesem Sinne de novo, s. o.) passt hingegen besser zur Schöpfungslehre, weil man eine programmierte Situation annehmen kann, in der Gene durch geringfügige Änderungen neue, bisher nicht beobachtete Funktionen aufweisen können. Nur in diesem funktionalen Sinne sind die Gene wirklich neu.
Literatur
Borger P (2020) De Novo – Gene aus dem Nichts? Interpretationsfehler oder komplexes Genom? Stud. Integr. J. 22, 88–96.
An NA et al. (2023) De novo genes with an lncRNA origin encode unique human brain developmental functionality. Nature Ecology & Evolution 7, 264–278, https://www.nature.com/articles/s41559-022-01925-6.
Brown W (2023) Study Finds Human Gene Linked to Larger Brains Arose from Non-Protein Coding (“Junk”) DNA. https://www.resonancescience.org/blog/study-finds-human-gene-linked-to-larger-brains-arose-from-non-protein-coding-junk-dna; aufgerufen am 14,08.2023)
Karger A et al (2018) Variation and Novelty in Evolution:De Novo Genes arise and enable Protein Structural Innovation. Intelligent Systems for Molecular Biology Conference, Chicago. https://www.youtube.com/watch?v=_1WNiXr2XqY, Min. 15:03 – 15:30.
Lau KF & Dill KA (1990) Theory for protein mutability and biogenesis, Proc. Natl. Acad. Sci. USA 87, 638–642.
Scholl B (2023) Beherrschen Schimpansen etwa doch Grammatik? W+W-Onlineartikel, vom 27.03.2023, https://www.wort-und-wissen.org/artikel/schimpansengrammatik/.
Wilson BA et al. (2017) Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nat. Ecol. Evol. 1, 0146, doi:10.1038/s41559-017-0146.
Autor dieserNews: Peter Borger
© 2023, http://www.genesisnet.info/schoepfung_evolution/n321.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
07.07.23 Photosynthese – wie Biologie von Physik profitiert
Photosynthese nutzt möglicherweise Quantenphänomen: Die Bose-Einstein-Kondensation
Organismen wie Bakterien, Algen und Pflanzen nutzen die Sonnenenergie, um ihre Biomasse aufzubauen. Dies erscheint uns geradezu selbstverständlich. Physikalische Phänomene wie Bose-Einstein-Kondensate hingegen gelten als exotisch. Kürzlich fanden Biophysiker heraus, dass es eine Verbindung zwischen diesen beiden so unterschiedlichen Phänomenen geben könnte und ermöglichten so einen tiefen Einblick in die Energieübertragung in Bio-Molekülen und Materialien in einem Lichtsammelkomplex, wie er in grünen Schwefelbakterien auftritt.
Die Bose-Einstein-Kondensation ist ein Quantenphänomen, bei dem eine große Anzahl von Bosonen* gleichzeitig den Grundzustand (d. h. das niedrigste Energieniveau) eines Systems einnimmt. Im Jahr 1925 von dem Quantenphysiker Satyendra Nath Bose vorhergesagt, wurden Bose-Einstein-Kondensate 70 Jahre später von Weimann, Cornell und Ketterle experimentell festgestellt, eine Entdeckung, wofür sie 2001 den Nobelpreis erhielten (Schwarz 2019).
*Bosonen (= Ein Teilchen mit ganzzahligem Eigendrehimpuls (Spin), z. B. Photonen)
Bose-Einstein-Kondensate, die auch als fünfter Zustand der Materie bezeichnet werden (neben fest, flüssig, gasförmig und plasmatisch), entstehen, wenn Atome auf nahezu den absoluten Nullpunkt abgekühlt werden. Die offensichtlichste Eigenschaft eines Bose-Einstein-Kondensats ist, dass ein großer Teil seiner Teilchen denselben, nämlich den niedrigsten, Energiezustand einnimmt. Bei im Labor erzeugten Bose-Einstein-Kondensaten kann dies durch Messung der Geschwindigkeitsverteilung der Teilchen bestätigt werden.
Es gibt auch die Bose-Einstein-Kondensation von Exzitonen*, wenn diese sich zu einem einzigen sog. kohärenten Quantenzustand, dem so genannten Exzitonenkondensat, verfestigen. Dieser Zustand ermöglicht einen verlustfreien Energietransfer, der jedoch typischerweise nur unter extremen Laborbedingungen in hoch geordneten Materialien stattfindet (Schouten 2023).
*Exzitonen (= Kombinationen aus einem Elektron und einem positiven Loch (ein leerer Elektronenzustand in einem Valenzband, das zuvor durch das Elektron besetzt wurde), das sich frei durch einen nichtmetallischen Kristall bewegen kann)
Bei der Photosynthese wird mithilfe des Chlorophylls* Wasser und Kohlendioxid durch Lichtenergie in pflanzliche Stoffe (Kohlenhydrate) umgewandelt. Dazu muss die Lichtenergie nach der Absorption eines Photons im photosynthetischen Reaktionszentrum weitergeleitet werden. (Das photosynthetische Reaktionszentrum ist ein Komplex aus mehreren Proteinen, Pigmenten und anderen Co-Faktoren, die zusammen die primären Energieumwandlungsreaktionen der Photosynthese durchführen; die Stelle in den photosynthetischen Molekülen, an der Photonen gesammelt werden.) Das ist aber eigentlich ein Problem. Denn die Atome, aus denen die komplexen molekularen Maschinen der Photosynthese bestehen, verhalten sich wie ein dichter Wald. Wie aber kann die Energie jemals das photosynthetische Reaktionszentrum erreichen, wenn sie im „molekularen Wald“ immer wieder auf Atome prallt? Das wäre so, als würde man mit einem Jagdgewehr in den Wald schießen und hoffen, ein Ziel zu treffen, obwohl Bäume im Weg stehen.
*Chlorophylls (= „Blattgrün“, einem Komplex aus Makromolekülen)
Doch den photosynthetischen Organismen gelingt es, das Sonnenlicht in Blätter, Stängel und Blüten umzuwandeln, als ob der atomare Wald nicht existierte. Wie machen sie das?
Eine neue Studie der Universität Chicago, die am 28. April 2023 in der Zeitschrift PRX Energy veröffentlicht wurde, legt nahe, dass dies durch ein Exzitonenkondensat ermöglicht wird, das dem unter extremen Bedingungen im Labor gefundenen sehr ähnlich ist. Es ermöglicht einen physikalischen Prozess, analog zu Supraleitung und Suprafluidität, der es der Energie ermöglicht, verlustlos durch ein Material zu fließen. Bildhaft gemäß dem obigen Gleichnis: Die Gewehrkugel kann Bäume durchdringen, ohne dabei Energie zu verlieren.
Dass Pflanzen vermutlich einen sehr ähnlichen Mechanismus wie bei der Bose-Einstein-Kondensation nutzen, war unerwartet und hat großes wissenschaftliches Interesse geweckt – denn was Wissenschaftler im Labor nur bei Temperaturen um den absoluten Nullpunkt erreichen können, leisten Pflanzen offenbar bei Raumtemperatur. Einer der leitenden Forscher, Prof. David Mazziotti, kommentierte: „Soweit wir wissen, waren diese Bereiche noch nie miteinander verbunden, daher fanden wir dies sehr interessant und aufregend“ (Lerner 2023). Und Anna Schouten, eine der Mitautorinnen, erklärt: „Die photosynthetische Lichtsammlung findet in einem [biologischen] System bei Raumtemperatur statt, dessen Struktur zudem ungeordnet ist – ganz im Gegensatz zu den ursprünglichen kristallisierten Materialien und den kalten Temperaturen, die man zur Herstellung von Exzitonenkondensaten verwendet“ (Lerner 2023).
Die beobachteten Kondensate sind zwar nicht so perfekt wie unter extremen Laborbedingungen, aber die Effizienz der Energieübertragung im photosynthetischen System wird fast verdoppelt (Schouten 2023).
Aber wie sind die Forscher zu dieser Entdeckung gekommen, da es keine Möglichkeit gibt, diese atomaren Wechselwirkungen mit bloßem Auge oder mithilfe von Mikroskopen zu sehen? Mazziottis Labor hat sich auf die Erforschung komplizierter atomarer und molekularer Wechselwirkungen mit Hilfe hochentwickelter Computermodelle spezialisiert, die ihnen einen Einblick in das zugrunde liegende Verhalten der Teilchen geben. In der Studie wollten sie herausfinden, was auf molekularer Ebene bei der Photosynthese passiert.
Wenn ein Photon von der Sonne auf ein Blatt trifft, löst es eine Veränderung in einem speziellen Molekül aus, dem so genannten Chromophor. Chromophore sind lichtabsorbierende Moleküle, die in der Regel an eine Proteinstruktur gebunden sind, die sie an Ort und Stelle hält. Die in dem Photon gebundene Energie regt ein Elektron des Chromophors an, wobei ein „Loch“ in dem Chromophor entsteht. Das Elektron und das „Loch“ können nun durch das Blatt wandern und die Energie des Photons zu einem anderen Bereich befördern. Dieses wandernde Elektronen-Loch-Paar wird als „Exziton“ bezeichnet. Als das Team um Mazziotti eine Gesamtübersicht erstellte und modellierte, wie sich mehrere Exzitonen bewegen, fiel ihnen etwas Merkwürdiges auf. Sie sahen Muster in den Bahnen der Exzitonen, die bemerkenswert vertraut aussahen (Lerner 2023).
Was die Forscher beobachteten, entsprach weitgehend dem Verhalten eines Materials, das als das bereits erwähnte Bose-Einstein-Kondensat bekannt ist. Die von den Lichtphotonen erzeugten Exzitonen verbinden sich zu einem einzigen Quantenzustand – ähnlich wie die Geiger in einem Orchester, die alle einen Ton perfekt gestimmt spielen. Im Quantenuniversum der Teilchenphysik kann sich die Energie so ohne Reibung durch das Material bewegen. Dieses Phänomen des energetisch verlustfreien Transports hat viel Aufmerksamkeit erregt, da es für hochempfindliche elektronische Geräte und Supercomputer interessant ist.
Es ist möglich, dass diese Entdeckungen bei photosynthetisierenden Bakterien, die bei Raumtemperatur arbeiten, neue Möglichkeiten für die Erzeugung synthetischer Materialien für zukünftige Technologien eröffnen. Im Labor ist ein perfektes ideales Exzitonenkondensat empfindlich und erfordert eine Menge spezieller Bedingungen. Systeme, die mit solchen in der Natur vorkommenden Materialien gebaut werden und bei Raumtemperatur das Gleiche tun, öffnen die Tür zu realistischeren technologischen Anwendungen. Somit könnte das ausgefeilte Design in der Natur wieder einmal als Vorbild für die Verbesserung der Technologie dienen – mit Anwendungen von Solarzellen bis hin zu Computern.
In dem Artikel wird nicht darüber spekuliert, wie diese „einfachen“ Schwefelbakterien den effizienten Energietransport „erfunden“ haben. „Die Evolution war es“, wird normalerweise geantwortet, um alles zu „erklären“, was in der Natur beobachtet wird, doch passt dies hier offensichtlich nicht.
Quellen
Lerner L (2023) Scientists find link between photosynthesis and ‚fifth state of matter‘. https://phys.org/news/2023-05-scientists-link-photosynthesis-state.html
Schouten AO et al. (2023) Exciton-Condensate-Like Amplification of Energy Transport in Light Harvesting. PRX Energy 2, 023002, https://journals.aps.org/prxenergy/abstract/10.1103/PRXEnergy.2.023002
Schwartz M (2019) Statistical Mechanics. https://scholar.harvard.edu/files/schwartz/files/12-bec.pdf
Autor dieserNews: Peter Borger
© 2023, http://www.genesisnet.info/schoepfung_evolution/n318.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
26.10.22 Gleiche Variationsmechanismen im Gehirn von Menschen und Kraken
Transposons („springende Gene“) aus der LINE-Familie sind genetische Elemente, die sich verdoppeln und Kopien von sich selbst im Genom hinterlassen können. Lange Zeit wurden sie als „Junk-DNA“ (Überbleibsel alter Viren) betrachtet, doch in jüngster Zeit hat sich gezeigt, dass sie für die Entwicklung und Komplexität des Nervensystems von Bedeutung sind. Völlig unerwartet sind dieselben springenden Gene sowohl im menschlichen Gehirn als auch im Gehirn von Kraken (Octopus) aktiv.
Mit * versehene Begriffe werden in einem Glossar am Ende des Artikels genauer erklärt.
Einleitung
Oktopusse (Echte Kraken) gehören zu den intelligentesten Tieren. Wie der Mensch verfügen sie über fortgeschrittene kognitive Fähigkeiten und Problemlösungskompetenzen, die für Neurobiologen von großem Interesse sind. Kein Wunder, dass sich die Forschung zunehmend mit der Erforschung des Gehirns und des Lernens dieser schlauen und neugierigen Tiere beschäftigt. Neurobiologen haben bereits früher festgestellt, dass das als LINE1 bekannte Transposon* in menschlichen Gehirnzellen aktiv ist (Upton et al. 2015). Man nimmt an, dass ihre Aktivität für Lernen und Kognition (Wahrnehmung, Erfahrung, Denken) entscheidend ist, da sie sich in der Nähe von Genen anreichern, die mit diesen Prozessen in Zusammenhang stehen. Jetzt haben Neurowissenschaftler dieselbe Familie von LINE-Transposons in den Neuronen von Kraken gefunden und sie auch mit deren kognitiven Fähigkeiten in Verbindung gebracht. Im Rahmen von Evolution wäre dies ein unglaublicher Fall von Konvergenz*.
Variation-inducing genetic elements (VIGE)
Ein großer Teil der Genome aller Organismen besteht aus Elementen, die Wissenschaftler heute als transponierbare und transponierte Elemente (TE) bezeichnen. Es handelt sich um DNA-Sequenzen, die von einer Stelle des Genoms zu einer anderen wandern und dabei Kopien von sich hinterlassen können. Die komplexesten TEs sind endogene Retroviren (kurz: ERV) und sog. „long interspersed nuclear elements“ (kurz: LINE). Ungefähr 8 % des menschlichen Genoms bestehen aus ERVs und 17 % aus LINEs. Sie wurden lange Zeit als Junk-DNA, als egoistische Gene und funktionslose evolutionäre Relikte betrachtet. Eine wachsende Zahl von Studien hat jedoch gezeigt, dass diese Elemente wichtige strukturelle und regulatorische Elemente des Genoms bilden; und sie werden zunehmend als eine wichtige treibende Kraft der Evolution eingeschätzt (Xing et al. 2007).
In früheren Arbeiten habe ich die LINE-Elemente als „variation-inducing genetic elements“ (VIGEs) bezeichnet (Terborg 2009a; 2009b; 2010; 2013), also als genetische Elemente, die Variation erzeugen. Damit ist gemeint, dass diese Elemente als Genschalter fungieren und/oder den genetischen Kontext so beeinflussen, dass die Genexpression in ihrer Umgebung beeinflusst wird. Durch ihr Vorhandensein können Gene mehr oder weniger aktiv sein, was zu genetischer Variation führt. Dass diese Bezeichnung treffend ist, wird nun auch durch die jüngste Hirnforschung deutlich. Sie zeigt, dass Transposons in hohem Maße zu Merkmalen beitragen, die mit der Komplexität des Nervensystems verbunden sind. Insbesondere die Retrotransposition* von LINE1 in Zellen des Hippocampus* wurde mit Kognition und Lernen in Verbindung gebracht (Muotri et al. 2005; Singer et al. 2011). Studien an der Fruchtfliege Drosophila haben ebenfalls gezeigt, dass Lernen und Gedächtnis mit der Aktivität von transponierbaren Elementen in Gehirnzellen zusammenhängen und dass ihre Aktivität fein reguliert ist (Kohlrausch et al. 2021; Protasova et al. 2021).
Konvergente Evolution?
Um festzustellen, ob im Gehirn von Kraken ein ähnlicher Mechanismus abläuft, hat eine Gruppe italienischer Wissenschaftler die sog. „Sequenzierungstechnologie der nächsten Generation“ eingesetzt, um die molekulare Zusammensetzung der Gene zu analysieren, die im Nervensystem des Gemeinen Kraken (Octopus vulgaris) und des Kalifornischen Kraken (Octopus bimaculoides) aktiv sind (Petrosino et al. 2022). Auf diese Weise konnten sie alle funktionellen TEs, einschließlich der LINE-Elemente, identifizieren und ihre genaue Position in der DNA bestimmen. Sie fanden heraus, dass die Aktivität eines der LINE-Elemente zu spezifischen Signalen in Neuronen führte, die zu Bereichen gehören, die mit Verhaltensplastizität in Verbindung gebracht werden, dem Gegenstück zum Hippocampus beim Menschen. Die Forscher kamen zu dem Schluss, dass „ein konvergenter evolutionärer Prozess, der die Aktivität von Retrotransposons* im Gehirn einbezieht, für die Evolution hochentwickelter kognitiver Fähigkeiten in dieser Gattung (Octopus) wichtig war“ (Petrosino et al. 2022).
Im System der Bilateria (Zweiseitentiere) könnten Mensch und Krake kaum weiter auseinander liegen: Kraken gehören zum Stamm der Weichtiere, der Mensch zu dem der Chordatiere. Im Rahmen des Evolutionsmodells sind die beiden Stämme durch etwa 500 Millionen Jahre evolutionärer Entwicklung getrennt. Dennoch haben Menschen und Kraken in einiger Hinsicht sehr ähnliche Augen – so genannte Kameraaugen –, die als Ergebnis konvergenter Evolution interpretiert werden. Als Konvergenz bezeichnet man die hypothetische Vorstellung, dass zwei (oder mehr) Organismen unabhängig voneinander sehr ähnliche biologische Strukturen evolutiv entwickelt haben. Nun müssen die Forscher eine weitere bemerkenswerte Konvergenz postulieren in Bezug darauf, wie beide Organismen durch den unabhängigen Erwerb von Transposons hochentwickelte kognitive Fähigkeiten erlangten.
Die Beobachtung, dass Menschen und Kraken genau den gleichen Mechanismus nutzen, um durch Retrotransposition von LINE-Variationen in Gehirnneuronen zu erzeugen, bedarf einer Erklärung. Wie ist es zwei unterschiedlichen Stämmen, Chordatieren und Weichtieren, gelungen, denselben Lernmechanismus unabhängig voneinander zu entwickeln? Die italienischen Forscher Giuseppe Petrosino und Stefano Gustincich kommentierten dies wie folgt:
„Diese Ähnlichkeit zwischen Mensch und Krake, die die Aktivität eines LINE-Elements im Sitz der kognitiven Fähigkeiten zeigt, könnte als ein faszinierendes Beispiel für konvergente Evolution erklärt werden – ein Phänomen, bei dem sich in zwei genetisch entfernten Arten derselbe molekulare Prozess unabhängig voneinander entwickelt, als Reaktion auf ähnliche Bedürfnisse“1 (Anonymus 2022).
Wenn beobachtet wird, dass zwei (oder mehr) verschiedene Organismen die gleichen oder sehr ähnliche Merkmale aufweisen, die nicht das Ergebnis von Abstammung sein können, muss man sich im Rahmen der Evolutionsbiologie auf konvergente Evolution berufen. Es sollte angemerkt werden, dass konvergente Evolution keine Erklärung ist, sondern ein Begriff für eine unerklärte Beobachtung, die Evolution voraussetzt. Haben die Autoren die Evolutionsprozesse untersucht, die zum heutigen Zustand geführt haben? Nein, sondern sie untersuchten Lernprozesse bei Kraken und fanden völlig unerwartet, dass dabei derselbe Mechanismus angewendet wird, der bereits beim Menschen bekannt war.
Das Spektakuläre daran ist, dass – evolutionär gesehen – an der Basis der Chordatiere ein Gehirn noch nicht existierte, wie wir bei den heutigen Stachelhäutern beobachten können. Wie kann es also zu einer evolutionären genetischen Konvergenz bei Arten kommen, deren gemeinsamer Vorfahre das entsprechende Organ, in dem es aktiv ist, gar nicht besaß? Eine ähnliche Frage kann für die LINE-Elemente gestellt werden. Sie müssen bereits vor der Aufspaltung der Weichtiere und Chordatiere vorhanden gewesen sein und später unabhängig voneinander in verschiedenen Linien für gleiche Zwecke kooptiert* worden sein. Das bedeutet, dass sie bereits vor der sog. Kambrischen Explosion in einem hypothetischen einfacher gebauten Vorfahren vorhanden gewesen sein müssen, um wesentliche biologische Funktionen zu erfüllen. Diese Funktionen konnten jedoch nicht die Gehirnzellen betreffen, da sich die Gehirne noch nicht entwickelt hatten.
LINEs sind keine Überbleibsel von Viren
Die evolutionäre Erklärung für die Vielzahl der ERVs und LINEs in den Genomen ist, dass es sich hierbei um die Überreste von Retroviren handeln soll, die vor Millionen von Jahren in die Genome eingedrungen sind. In Anbetracht der neuen Daten bei den Kraken müsste dies für die LINE-Elemente noch vor der Kambrischen Explosion geschehen sein. Seitdem sollen diese Gene überall im Genom Kopien und funktionslose Bruchstücke von sich hinterlassen haben. Einige moderne Retroviren, wie das humane Immundefizienz-Virus (HIV), ähneln tatsächlich ERVs. Alternativ zu der Vorstellung, dass ERVs als integrierte Überbleibsel von (HIV-ähnlichen) Retroviren gedeutet werden, lassen sich Retroviren (einschließlich HIV) besser als transformierte ERVs erklären, die sich aus dem Genom losgelöst haben (Borger 2009b; 2010; 2013). Die Annahme, dass es sich bei LINEs um Überbleibsel von Retroviren handelt, wird durch die empirischen Daten nicht gestützt, denn im Gegensatz zu ERVs gibt es keine Retroviren, die LINEs ähneln. Dies lässt sich am Beispiel von LINE1, einem im menschlichen Genom aktiven transponierbaren Element, verdeutlichen. LINE1 ist ein komplexes genetisches Element mit zwei offenen Leserahmen*: ORF1 und ORF2. Das von ORF2 codierte Protein ermöglicht wesentliche enzymatische Aktivitäten für die reverse Transkription sowie für die Integration einer neu transponierten Kopie von LINE1. Somit pflanzt sich LINE1 durch einen Copy-Paste-Mechanismus fort und hinterlässt dabei identische Kopien an verschiedenen Stellen im Genom.
Die genaue Rolle von ORF1 ist noch ungeklärt. Es codiert ein Protein mit proteinbindenden Eigenschaften, kann aber auch als Nukleinsäure-Chaperon* fungieren (Upton et al. 2011). Der Ursprung von LINE1 ist jedoch völlig unbekannt. Der einzige Grund, LINE als Überbleibsel von Retroviren zu betrachten, besteht darin, dass sie ein Reverse-Transkriptase-Gen besitzen, das dem von ERVs geringfügig ähnelt. Es sollte jedoch betont werden, dass LINEs einen einzigartigen genetischen Aufbau haben, der sie zu einzigartigen genetischen Elementen macht, die nicht mit Retroviren verwandt sind. Die italienischen Hirnforscher liefern nun weitere Beweise dafür, dass sie in Genomen als variationsinduzierende genetische Elemente wirken. Dass sie in einer fernen Vergangenheit als Viren entstanden sind, ist lediglich eine Vermutung ohne empirische Belege.
Es bleibt rätselhaft, wie die LINE-Elemente über Hunderte von Millionen von Jahren erhalten und konserviert werden konnten, wenn es sich lediglich um virale Invasionen handelte, die durch sich anreichernde Mutationen ziemlich schnell zerstört werden könnten, wenn sie nicht sofort wichtige Funktionen ausüben. Die Konvergenz durch unabhängige Kooption von LINE übersteigt nach aktuellem Kenntnisstand die Glaubwürdigkeit einer evolutionären Entstehung.
Argument gegen die gemeinsame Abstammung
Seit über einem Jahrhundert wird die Evolutionsbiologie vom neodarwinistischen Forschungsprogramm dominiert. Die Haupthypothese dieses Programms besagt, dass alle Arten durch natürliche Prozesse (Selektion von Zufallsmutationen) aus einem einzigen gemeinsamen Vorfahren hervorgegangen sind. Im Rahmen der Schöpfungslehre hingegen wird die unabhängige Erschaffung von Urtypen (oder Grundtypen) mit eingebauten flexiblen Genomen postuliert, die sich verändern und anpassen und neue Arten hervorbringen können (Terborg 2008). Demnach enthielten die Genome der Urtypen von Anfang an eine begrenzte Anzahl von VIGEs, einschließlich ERVs und LINEs. In verschiedenen Urtypen könnten sich VIGEs an genau derselben Stelle in der DNA befunden haben, was dann erklärt, warum manche VIGEs in den Genomen moderner Organismen an derselben Stelle zu finden sind, unabhängig von der Annahme einer gemeinsamen Abstammung.
Die Funktionalität von LINEs ist sehr wichtig, die Plausibilität des Evolutions- und des Schöpfungsansatzes zu beurteilen. Wären LINEs ohne Funktion und würden sie sich zufällig in Genome einfügen, würde bei Vorliegen von ähnlichen Positionen der LINEs die Annahme der gemeinsamen Abstammung im neodarwinistischen Rahmen stark unterstützt (vgl. Jorritsma 2022). Wären die LINEs hingegen funktionell und würde ihre genomische Integration stark reguliert und kontrolliert, wäre das Argument für die gemeinsame Abstammung schwach. Das Vorhandensein der gleichen LINEs (und anderer VIGEs) am gleichen Ort in den Genomen verschiedener Arten würde dann lediglich auf ein „verschachteltes Hierarchie-Argument“ hinauslaufen, das auch im Rahmen von Schöpfung verstehbar ist.
Die Tatsache, dass wir sowohl bei Wirbeltieren als auch bei Weichtieren LINEs mit genau derselben Funktion finden, ist ein starkes Argument dafür, dass gemeinsame Retrotransposons, selbst wenn sie sich an genau derselben Stelle in der DNA befinden, nicht unbedingt auf eine gemeinsame Abstammung schließen lassen. Vielmehr spricht ihre Funktionalität für ein vorgelagertes modulares Designsystem, das kontrollierte und regulierte Variationen hervorruft (Borger 2009b; 2010; 2013).
Anmerkung
1 Das ist teleologisches Denken: „Bedürfnisse“ implizieren ein Ziel, das es im Rahmen von Evolution nicht geben darf.
Glossar
Hippocampus: Gehirnregion, die zum limbischen Kortex (Limbisches System) gehört und als Schaltstelle zwischen dem Kurz- und dem Langzeitgedächtnis fungiert. Der Hippocampus ist einer der wenigen Bereiche im Gehirn, in dem ein Leben lang neue Nervenzellen gebildet werden können.
Konvergenz: In der Evolutionsbiologie eine strukturelle, physiologische oder verhaltensmäßige Ähnlichkeit, die auf gleicher Funktion beruht und unabhängig entstanden ist.
Kooption: Übernahme von Merkmalen (oder Genen) in einen neuen Funktionszusammenhang.
LINE: Abkürzung von long interspersed nuclear element. à Retrotransposon, das in höheren Lebewesen vorkommt.
Nukleinsäure-Chaperon: Proteine, die in Zellen DNA oder RNA begleiten und/oder transportieren.
Offener Leserahmen (engl. open reading frame; ORF): DNA-Sequenz zwischen Start- und Stoppcodon, die die Aminosäuresequenz eines Proteins oder die Nukleotidsequenz eines RNA-Moleküls codiert.
Retrotransposon: Eine DNA-Sequenz, das sich im Genom über einen Copy/Paste-Mechanismus vermehrt. Es kopiert sich selbst über ein RNA-Molekül, das dann umgewandelt und in die DNA integriert wird. Auf diese Weise kann es vererbbare Variationen hervorrufen.
Transposon (pl: Transposons oder Transposonen): Genetisches Element (DNA- Sequenz), das sich im Genom umlagern und somit neue genetische Zusammenhänge erzeugen kann. Transposons werden derzeit als wichtige Triebkräfte für Anpassungen auf mikroevolutiver Ebene angesehen, aber auch als Triebkräfte für die großen phänotypischen Veränderungen, die für die Makroevolution erforderlich sind. Es sollte beachtet werden, dass sie dem Genom keine neuen genetischen Informationen hinzufügen, sondern bereits vorhandene genetische Programme freisetzen können.
VIGE: Abkürzung von variation-inducing genetic element. Eine DNA-Sequenz, die Variation erzeugen kann. Damit ist gemeint, dass diese Elemente als Genschalter fungieren und/oder den genetischen Kontext so beeinflussen, dass die Genexpression in ihrer Umgebung beeinflusst wird. Durch ihr Vorhandensein können Gene mehr oder weniger aktiv sein, was zu genetischer Variation führt.
ERV: Abkürzung von endogenous retrovirus. Transposon, das in höheren Lebewesen vorkommt.
Verschachtelte Hierarchie: Im System der Lebewesen beobachtetes Ordnungsprinzip, das durch eine abgestufte Rangordnung charakterisiert ist, d.h. Gruppen in größeren Gruppen innerhalb von noch größeren Gruppen.
Quellen
Anonymus (2022) Study: Same ‘Jumping Genes’ are Active in Octopus and Human Brains. Sci News, Jun 28, 2022, https://www.sci.news/genetics/octopus-human-brain-transposable-elements-10943.html
Jorritsma R (2022) How Well Does Evolution Explain Endogenous Retroviruses? – A Lakatosian Assessment. Viruses 14, https://doi.org/10.3390/v14010014
Kohlrausch FB, Berteli TS, Wang F, Navarro PA & Keefe DL (2021) Control of LINE-1 Expression Maintains Genome Integrity in Germline and Early Embryo Development. Reproductive Sciences 2021, 1–13.
Muotri AR, Chu VT, Marchetto MCN et al. (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 2005 435, 903–910.
Petrosino G, Ponte G, Volpe M et al. (2022 )Identification of LINE retrotransposons and long non-coding RNAs expressed in the octopus brain. BMC Biol 20, 116, https://doi.org/10.1186/s12915-022-01303-5
Protasova MS, Andreeva TV & Ivanovich Rogaev EI (2021) Factors Regulating the Activity of LINE1 Retrotransposons. Genes (Basel) 12, 1562.
Singer T, McConnell MJ, Marchetto MCN et al. (2011) LINE-1 Retrotransposons: Mediators of Somatic Variation in Neuronal Genomes? Trends in Neuronal Sciences 33, 345–354.
Terorg P (2008) The design of life: part 3 – baranomes. J. Creation 22, 68–76.
Terborg P (2009a) The design of life: part 3 – an introduction to variation-inducing genetic elements, J. Creation 23, 99–106.
Terborg P (2009b) The design of life: part 4 – variation-inducing genetic elements and their functions. J. Creation 23, 107–114.
Terborg P (2013) The ‘VIGE-first hypothesis’ – how easy it is to swap cause and effect. J. Creation 27, 105–112.
Terborg P (2010) ERVs and LINEs – along novel lines of thinking. J. Creation 32, 8–10.
Upton KR, Baillie JK & Faulkner GJ (2011) Is somatic retrotransposition a parasitic or symbiotic phenomenon? Mob. Genet. Elements 1, 279–282.
Upton KR, Gerhardt DJ, Jesuadian JS et al. (2015) Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161, 228–239.
Xing J, Witherspoon DJ, Ray DA, Batzer MA & Jorde LB (2007) Mobile DNA elements in primate and human evolution Am. J. Phys. Anthropol. Suppl 45, 2–19.
Autor dieserNews: Peter Borger
© 2022, http://www.genesisnet.info/schoepfung_evolution/n307.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
12.04.22 Epigenetik und programmierte Anpassungen
Hin und wieder wird von Organismen berichtet, deren Nachkommen auf neue Umweltprobleme reagiert haben. Häufig wird dies als „schnelle Evolution“, „rasante Evolution“ oder ähnliches bezeichnet. Ein besserer Begriff wäre „programmierte Anpassung“, denn es ist das Genom (Erbgut) selbst, das auf neue Herausforderungen vorbereitet ist und es den Organismen ermöglicht, sich an sie anzupassen. Diese außergewöhnliche Fähigkeit wird heute unter dem Begriff „Epigenetik“ gefasst und bezieht sich auf erbliche Veränderungen, die nicht auf Mutationen in der DNA-Sequenz zurückgeführt werden können. Der Begriff Epigenetik wurde eingeführt, um alle Prozesse zu erfassen, die auf die Genaktivität Einfluss nehmen, ohne dass dabei die DNA-Sequenz verändert wird. Die epigenetischen Veränderungen können sogar auf Tochterzellen übertragen werden. Seit fast einem Jahrhundert, nachdem der Begriff zuerst auftauchte, fängt man an zu verstehen, wie die Körper der höheren Organismen sich mittels der Epigenetik aus einer einzigen Zelle entfalten können.
Schmetterlinge schlüpfen als Raupen aus ihren Eiern. Aus den sechs kurzen Beinen der Raupe müssen sechs lange, elegante Schmetterlingbeine werden. Ihre Kiefern, mit denen die Blätter zerkauen, müssen durch den langen nektarsaugenden Rüssel ersetzt werden, und die einfachen Raupenaugen werden in Facettenaugen umgewandelt. Die Flügel – Organe, die die Raupe noch nie zuvor hatte – sollten ebenfalls ausgebildet werden. Darüber hinaus hat der erwachsene Schmetterling ein völlig neues Atmungssystem, das aus einem von außen in den Körper verzweigendes Luftröhrensystem besteht, das einen komplett neuen Brustkorb und ein neues Abdomen mit einem neuen Verdauungstrakt versorgt.
Diese Metamorphose ist ein Wunder. Das eine im Ei vorhandene Genom (und Epigenom, s. u.) mit seiner DNA-Sequenz einschließlich aller Modifikationen enthält zwei völlig unterschiedliche genetische Baupläne, von denen einer für eine Raupe verwendet wird, während der andere Informationen darüber enthält, wie der Schmetterling aussieht. Diese völlig unterschiedlichen genetischen Anteile an den Bauplänen von Raupe und Falter sollten natürlich nicht gleichzeitig aktiv sein, denn dann würde ein nicht lebensfähiges Ungetüm entstehen. In der Raupe muss der Bauplan des Schmetterlings unterdrückt werden, im Schmetterling muss der Bauplan der Raupe ausgeschaltet bleiben. Um dies zu erreichen, verpuppt sich das Tier und während der Puppenphase wird der Raupenbauplan langsam abgeschaltet und der des Schmetterlings aktiviert.
Die Metamorphose der Schmetterlinge ist ein sehr anschauliches Beispiel für Epigenetik. Wir finden umfangreiche epigenetische Prozesse aber auch in allen mehrzelligen Organismen, die sich aus verschiedenen ausdifferenzierten Zell- und Gewebetypen zusammensetzen.
Epigenetische Markierungen
Grundsätzlich enthalten alle Zellen von mehrzelligen Organismen annähernd die gleiche Menge an Genen. Ein Gen ist nur dann brauchbar, wenn es seine Funktion in den richtigen Zellen im richtigen Augenblick und in den richtigen Verhältnisse, ausübt. Dazu ist in jedem Zelltyp, in jedem Gewebetyp und in jedem Organ nur ein Bruchteil der totalen Anzahl der Gene erforderlich. Tatsächlich sind nur einige hunderte – maximal einige Tausende – Gene pro Zelle aktiv. Eine differenzierte, ausgewachsene Leberzelle benötigt nur leberspezifische Gene, etwa 5% des Genoms. Ebenso braucht das Herz nur herzspezifische Gene, während T- und B-Zellen nur einen Bruchteil der Gene des Immunitätssystems verwenden. Wie steuern die Gewebezellen eine spezifische Anwendung der richtigen genetischen Programme?
Die Antwort ist: durch epigenetische Markierungen, d. h. funktionale Einheiten über der DNA-Sequenz. Es handelt sich um kleine Moleküle, die auf bestimmte Chromosomenabschnitte angehängt werden und auf diese Weise als genetische Schalter wirken, in dem sie Gene stummschalten oder aktivieren können. Epigenetische Markierungen weisen darauf hin, dass es in der Systemhierarchie der Genexpression eine übergeordnete Instanz gibt, die bestimmt, welche genetischen Programme verschlossen bleiben und welche zugänglich sind. Sie regulieren die Entwicklung und die Differenzierung der Zellen, indem sie die jeweils benötigen biologischen Programme zielgenau an- und abschalten können, und bedingen somit, dass aus einer Eizelle mehrere Hundert unterschiedliche Zelltypen entstehen.
Die wichtigsten epigenetischen Markierungen werden durch winzige biochemische Abänderungen hervorgerufen. In den Zellen ist eine Familie von Enzymen aktiv, die DNA-Methyltransferasen, die eine Methylgruppe, ein kleines, organisches Molekül, an vorbestimmte DNA-Sequenzen im Genom heften können. Durch die Ankupplung einer Methylgruppe wird diese Stelle im Genom gleichsam verriegelt, d. h. die genetische Information ist nicht mehr zugänglich und kann nicht mehr verwendet werden. Gene, die in einer bestimmten Zelle nicht notwendig sind, werden auf diese Weise inaktiviert und ausgeschaltet. Die Methylgruppen können von spezifischen Enzymen wieder entfernt werden, damit das genetische Programm wieder zugänglich und lesbar wird, falls das nötig wäre. Die DNA-Methylierung ist eine der bestgeklärten epigenetischen Mechanismen, die Zellen zur Verfügung haben, um organspezifische Gene ein- und auszuschalten.
Histoncode
Darüber hinaus kontrolliert auch der sogenannte Histoncode den Zugang zur genetischen Information. Histone sind Proteine, um die die Chromosomen (und damit DNA-Moleküle) geschlungen werden. Sie besitzen alle eine Domäne (Teil eines Proteins), die als Ausbuchtung aus den Proteinen hervorragt. Diese Domäne, oft bezeichnet als der Histonschwanz, ist aus Aminosäuren aufgebaut und kann, wie bei DNA-Methylierungen, mittels der Ankupplung chemischer Gruppen markiert werden. Die chemischen Markierungen der Histone bestehen aus Phosphatgruppen, Methyl- und Acetylgruppen werden durch Enzyme übertragen und bewirken, dass die Chromatinstruktur der Chromosomen lockerer (Euchromatin) oder kompakter (Heterochromatin) wird. Eine lockere Struktur vereinfacht die Transkription (Ablesung der Gen-Information) und erhöht die Genaktivität, während eine kompakte Struktur das Gegenteil bewirkt. Somit können diese epigenetischen Mechanismen die Transkription einzelner Gene oder ganzer Gruppen von Genen beeinflussen.
Die epigenetischen Markierungen eines Genoms beeinflussen auch die Art und Weise, wie sich das Chromatin in eine dreidimensionale Form faltet. Diese komplexe Faltung beeinflusst wiederum auch die Aktivität von Genen, indem sie verhindert, dass die Transkriptionsmaschinerie auf DNA-Regionen zugreift, die im Inneren des Genoms versteckt sind. Auf diese Weise können Zellen, die alle die gleiche DNA-Sequenz besitzen, zu verschiedenen Zelltypen werden. Der epigenetische Code enhält also Informationen, die der Zelle die Auswertung der genetischen Information ermöglichen, damit unterschiedlichen Organe, Gewebe und Zellen gebildet werden können.
Einflüsse auf die epigenetischen Markierungen
Interessante Beispiele sind Studien, die zeigen, dass die epigenetische Markierung, und somit das Ein- und Ausschalten von Genen, durch Nahrung und Lebensweise (z. B. extreme Stressfaktoren) beeinflusst werden kann. Dies geschieht durch einen unmittelbaren Einfluss der Umgebung auf die epigenetischen Instruktionen, die der DNA und den Histonen vermittelt werden. Die dadurch entstandenen Markierungen können sogar von den Eltern über einige Folgegenerationen vererbt werden.
Bei Pflanzen ist es gut belegt, dass die Eigenschaften der Nachkömmlinge epigenetisch so gesteuert werden können, dass sie auf die Lebensbedingungen vorbereitet sind, denen die Mutterpflanzen selbst ausgesetzt waren. Das ist beispielsweise der Fall, wenn die nächste Generation einer bestimmten Pflanzenart vor Raupenbefall geschützt werden soll. Dieser Schutz wird dadurch verbessert, dass die Dichte der Trichome (haarähnliche Strukturen auf der Pflanzenepidermis) erhöht wird. Als Reaktion auf Raupenfraß reagieren die wilden Rettich-Pflanzen (Raphanus raphanistrum) sofort mit einer Erhöhung der Dichte der schützenden Trichome. Wenn eine Raupe das erste Blatt beschädigt, nimmt die Dichte der Trichome vom dritten bis zum siebten Blatt zu. Dadurch verringerte sich die Häufigkeit der Raupenbesuche auf neuen Blättern und folglich auch der Raupenfraß. Das Gleiche wurde bei der Gauklerblume (Mimulus guttatus) in Kalifornien beobachtet. Hier vererben die geschädigten Mutterpflanzen eine höhere Trichomdichte an die Nachkommenschaft im Vergleich zur Nachkommenschaft von unbeschädigten Pflanzen. Der Nachwuchs ist so im Vorfeld besser gegen die pflanzenfressenden Raupen geschützt, wenn es zu einem weiteren Befall kommt. Dies ist ein klarer Fall von Vererbung eines epigenetisch erworbenen Merkmals. Vergleichbare generationsübergreifende Anpassungseffekte wurden bei der Widerstandsfähigkeit gegen Dürreperioden, gegen hohe und niedrige Temperaturen und sogar bei der Resistenz gegen Virusinfektionen beobachtet (Herman et al. 2011).
Molekularbiologische Beobachtungen belegen ebenso, dass die Kopfverzierung bei Käfern unter modulierbarer epigenetischer Kontrolle erfolgt. Manche männlichen Käfer zeigen große auffällige Ornamente oder eine Hörner, die das evolutive Ergebnis sexueller Selektion sein sollen. Allerdings sind die Größe und das Ausmaß, in dem sich diese Strukturen in einem Individuum entwickeln können, variabel und von der Ernährung abhängig. 2016 berichtete eine japanische Forschungsgruppe, dass die Ernährungswirkung auf die Größe der Ornamente des breitgehörnten Mehlstampfers (Gnatocerus cornutus) durch epigenetisch modifizierende Faktoren vermittelt wird. Wenn eine der Histon-Deacetylasen (HDAC1) in den Käferlarven ausgeschaltet wurde, schrumpften die Kiefern der nachfolgenden erwachsenen Tiere, während die Dämpfung der Expression eines anderen Enzyms (HDAC3) zu einer übertriebenen Ausprägung von Mundwerkzeugen führte, ohne andere Körperteile zu beeinflussen (Ozawaa et al. 2016).
Epigenetik beim Menschen
Ebenso wie bei den Pflanzen und Insekten gibt es auch beim Menschen eine Vererbung epigenetischer Information, der durch die Lebensweise der Mutter, des Vaters oder sogar durch die der Großeltern festgelegt wurde. So kann beispielsweise ein geringer Wuchs von Babys rauchender Mütter auf epigenetische Änderungen zurückgeführt werden, namentlich auf die veränderten Methylierungsmuster der DNA. Eine niederländische Studie legt nahe, dass die Enkelkinder hungernder Großeltern immer noch denselben epigenetischen Code wie ihre Großeltern erworben haben. Wiewohl noch immer nicht alle Einzelheiten bekannt sind, ist es klar, dass die Lebensweise und die Umgebung Einfluss auf die Eigenschaften des Nachwuchses ausüben.
Epigenetische Modifikationen erklären auch, wie die berühmten Darwinfinken als Reaktion auf plötzliche Umweltveränderungen wie Dürre oder Nässe sich so schnell anpassen können. Ein beachtliches Teil der Variation, die Darwin bei den Finken auf den Galapagos-Inseln beobachtete, wird heutzutage als umkehrbare epigenetische Veränderung erkannt (McNew et al. 2017). Diese Variation ist unabhängig von DNA-Mutationen und erfordert nur, dass die Information in der DNA auf eine andere Weise ausgeprägt wird.
Wenn neue Phänotypen nicht nur auf Mutationen in der DNA zurückzuführen sind, sondern auf reversible epigenetische Anpassungen, wie unterscheiden wir dann zwischen Evolution und programmierter Anpassung? Das kann nur durch detaillierte genetische Untersuchungen geklärt werden. Es ist jedenfalls möglich, dass Beispiele schneller Änderungen, die als Belege für Evolution interpretiert wurden, auf epigenetische Programmierung zurückzuführen sind. Programmierung ist aber ein Hinweis auf Planung und Voraussicht und somit auf einen Schöpfer.
Quellen
Herman JJ & Sultan SE (2011) Adaptive transgenerational plasticity in plants: case studies, mechanisms, and implications for natural populations. Front. Plant Genet. Genomics 2, 10-25.
Ozawaa T, Mizuharaa T, Aratab M, Shimadac M, Niimid T, Okadae K, OkadacY & Ohtaa K (2016) Histone deacetylases control module-specific phenotypic plasticity in beetle weapons. Proc. Natl. Acad. Sci. 113, 15042–15047.
McNew SM, Beck D, Sadler-Riggleman I, Knutie SA, Koop JAH, Clayton DH & Skinner MK (2017) Epigenetic variation between urban and rural populations of Darwin’s finches. BMC Evol. Biol. 17(1):183. doi: 10.1186/s12862-017-1025-9.
Autor dieserNews: Peter Borger
© 2022, http://www.genesisnet.info/schoepfung_evolution/n300.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
15.03.22 Mutationen sind doch nicht so zufällig
Eine langjährige Lehrmeinung unter Evolutionsbiologen war, dass Mutationen überall in einem Genom mit gleicher Wahrscheinlichkeit auftreten können. Befunde der Genanalytik zeigen jedoch, dass Mutationen nicht gleichverteilt sind und somit die Positionen, an denen Mutationen auftreten, nicht durchweg zufällig sind. Neue Forschungen an der Modellpflanze Acker-Schmalwand (Arabidopsis thaliana), über deren Erbgut sehr viel geforscht wird, bestätigen nun, dass Mutationen nicht gleichmäßig über das Genom verteilt sind. Eine im Januar 2022 in Nature veröffentlichte Studie belegt, dass Mutationen in Regionen des Genoms mit essenziellen Funktionen weniger häufig auftreten. Die zunächst angenommene Hypothese der Gleichverteilung muss also modifiziert werden: Nicht nur der Zufall, sondern im Genom vorhandene genetische Mechanismen sind dafür entscheidend, wo Mutationen vorgefunden werden.
Nach gängiger Lehrmeinung sind Mutationen 1. nicht vorhersehbar und hängen 2. nicht mit dem Verhalten, dem Lebensstil oder den Umweltbedingungen des Organismus zusammen. Diese Sichtweise wird von Futuyma (2005, 178f.) in seinem internationalen Standard-Lehrbuch wie folgt zusammengefasst:
„Mutationen sind in zweierlei Hinsicht zufällig. Erstens: Obwohl wir die Wahrscheinlichkeit vorhersagen können, dass eine bestimmte Mutation auftreten wird, können wir nicht vorhersagen, welche von einer großen Anzahl an Genkopien die Mutation durchlaufen wird. Zweitens … ist Mutation zufällig in dem Sinne, dass die Wahrscheinlichkeit, dass eine bestimmte Mutation auftritt, nicht davon beeinflusst wird, ob sich der Organismus in einer Umgebung befindet, in der diese Mutation vorteilhaft wäre, oder ob das nicht der Fall ist.“
Im Gegensatz zu dieser Lehrmeinung wurden in mehreren Studien sogenannte adaptive Mutationen nachgewiesen, also passende Mutationen als Reaktion auf Umweltveränderungen (Spetner 1997; Caporale 2003). Außerdem ist bekannt, dass ein hoher Prozentsatz der Mutationen an sogenannten DNA-Hotspots aufzutreten scheint, d. h. an Stellen in der DNA, die eher Mutationen zulassen (Terborg 2010; Borger 2019). Eine neue Studie (Monroe et al. 2022) bestätigt nun, was in der Schöpfungsforschung schon länger vermutet wurde, nämlich, dass einer der Hauptgrundsätze der Evolutionstheorie – die Zufälligkeit des Mutationsgeschehens – nur teilweise zutrifft.
Mutationen können in allen Zelltypen eines Organismus auftreten, also in den Gewebezellen und in den Zellen der Keimbahn (Spermien und Eizellen). Nur die Mutationen, die die Keimbahn betreffen, können an die Nachkommen weitergegeben werden, und diese sollen die Evolution vorantreiben. Im naturalistischen Rahmen der Evolutionstheorie sind alle Mutationen, auch die Mutationen der Keimbahn zufällig und Evolution verläuft daher ungerichtet. Durch natürliche Auslese sollen die zufälligen Mutationen selektiert („ausgewählt“) und fixiert werden, was im Trend zu besserer Anpassung und einen erhöhten Fortpflanzungserfolg der Nachkommen führen soll.
Möglicherweise haben die meisten Wissenschaftler aus diesem Grund bislang der Nicht-Zufälligkeit von Mutationen kaum Beachtung geschenkt (Borger 2019). Die wenigen Mutationen, die deutliche Anzeichen von Nicht-Zufälligkeit aufwiesen, wurden als Ausnahmen betrachtet, da sie nicht zur Theorie passten.
Das größte Hindernis für eine umfassende Untersuchung von Mutationen auf der Gen-Ebene war der Mangel an Daten und Analysemöglichkeiten. Das änderte sich mit den modernen Sequenzierungstechnologien und Computerkapazitäten mit entsprechenden Algorithmen, die es ermöglichen, Mutationen in großer Zahl zu untersuchen.
Der Modellorganismus Acker-Schmalwand
Die Acker-Schmalwand (Arabidopsis thaliana) ist ideal für genetische Studien, da sie ein kleines Genom besitzt (120 Millionen DNA-Buchstaben; zum Vergleich: Das menschliche Genom besitzt 3 Milliarden). Außerdem hat sie eine sehr kurze Generationszeit von 5 bis 6 Wochen, so dass Mutationen in den Nachkommen schnell entdeckt und untersucht werden können. Zudem gibt es für die Acker-Schmalwand reichhaltige Informationen über Sequenz- und Epigenom-Merkmale (d. h. Merkmale, die Meta-Informationen über die Gene enthalten, die z. B. markieren, ob bzw. wann Gene an- und abgeschaltet werden müssen).
Genome bestehen aus genreichen und genarmen Bereichen, die oft als „Genregionen“ bzw. „Genwüsten“ bezeichnet werden. Genregionen sind Abschnitte des Genoms, in denen sich die Gene befinden, einschließlich der sogenannten essenziellen Gene, die absolut unverzichtbar sind. Gene enthalten sehr spezifische Informationen, die für die Herstellung von Proteinen oder von RNA benötigt werden; sie sind relativ empfindlich gegen Mutationen und können daher durch Mutationen ihre Funktion leicht verlieren. Die Elemente der „Genwüsten“ dagegen können Mutationen besser abpuffern: Mutationen führen bei ihnen meist nicht unmittelbar zum Verlust ihrer Funktion. Diese Bereiche sind allerdings nicht funktionslos, die betreffenden Abschnitte enthalten verschiedene Elemente, die die räumliche Anordnung der Chromosomen bestimmen oder die zur Variation in den Nachkommen beitragen.
In ihrer Studie testeten Monroe et al. (2022) die Zufälligkeit von Mutationen, indem sie untersuchten, ob neue Mutationen gleichmäßig auf DNA-Regionen mit Genen und solche ohne Gene verteilt waren. Um die Mutationsrate und -position zu bestimmen, kultivierten die Forscher mehrere Generationen der Pflanzen über mehrere Jahre hinweg. Danach isolierten und sequenzierten sie die DNA von 1.700 Genomen und lokalisierten mehr als 1 Million Mutationen. Dabei stellten sie fest, dass die Teile der Genome, die Gene enthalten, eine viel geringere Mutationsrate aufwiesen als die nichtgenetischen Regionen. Die Mutationshäufigkeit war innerhalb von Gen-Regionen um die Hälfte, und in essenziellen Genen um zwei Drittel reduziert (Monroe 2022).
Einer der Autoren der neuen Studie, Grey Monroe, Pflanzengenetiker an der Universität von Kalifornien, kommentierte: „Ich war völlig überrascht von den nicht-zufälligen Mutationen, die wir entdeckt haben. Seit dem Biologieunterricht in der Schule hat man mir immer gesagt, dass Mutationen zufällig sind“ (zit. in BAKER 2022).
Mutationen treten also bei essenziellen Genen weniger häufig auf. Das Phänomen konnte nicht auf natürliche Selektion zurückgeführt werden, da die Pflanzen unter Laborbedingungen gezüchtet wurden und keine speziellen Selektionsdrücke wirksam waren. Das nicht zufällige Muster der Mutationen bei Gen- und Nicht-Gen-Regionen der DNA deutet darauf hin, dass es einen genetischen Mechanismus gibt, der mindestens einen Teil potenziell katastrophaler Mutationen verhindert. Aber wie könnte ein solcher Mechanismus funktionieren?
Wie werden potenziell schädliche Mutationen verhindert?
Die Forscher fanden heraus, dass essenzielle Gene spezielle Signale an DNA-Reparaturproteine aussenden, durch die sich selbst zu schützen können. Diese Signale werden nicht von der DNA selbst hervorgerufen, sondern von Histonen, speziellen Proteinen, um die sich die DNA wickelt, und so die Chromosomen bilden. Diese Signale gehören zu den Meta-Informationen des Epigenoms. „Basierend auf den Ergebnissen unserer Studie haben wir herausgefunden, dass Genregionen, insbesondere für die biologisch wichtigsten Gene, mit bestimmten chemischen Markierungen um Histone gewickelt sind“, so Monroe. „Wir vermuten, dass diese chemischen Markierungen als molekulare Signale wirken, um die DNA-Reparatur in diesen Regionen zu fördern“ (zit. in Baker 2022).
Diese chemischen Marker bzw. Signale sind nichts anderes als ein Code zur Stabilisierung wichtiger genetischer Information, damit der Organismus ohne Störungen funktionieren kann. Frühere Studien über Mutationen bei Krebspatienten haben ebenfalls ergeben, dass Histon-Proteine einen Code für DNA-Reparaturproteine tragen können, damit letztere Mutationen erkennen und reparieren können. Dies ist jedoch die erste Studie, die zeigt, dass ein solcher Histon-assoziierter Code das genomweite Mutationsmuster beeinflusst.
Ein codierter „Abwehrmechanismus“, der Mutationen von wichtigen Regionen fernhält, ist das, was man von einem vorausschauenden Designer erwartet, nicht jedoch von einem absichtslosen Prozess der Evolution. Es sei daran erinnert, dass die Nicht-Zufälligkeit von Mutationen nach bisher gängigen Evolutionstheorien nicht zu erwarten war. Somit ist die Frage berechtigt, ob diese neuen Erkenntnisse wichtige Teilaspekte bisheriger Evolutionstheorien in Frage stellen oder gar widerlegen. Diese Frage wird allerdings nicht gestellt. Es scheint vielmehr keine Rolle zu spielen, was wir beobachten, die Evolutionstheorie ist immer richtig: „Die Studie zeigt nur, dass diese genetischen Veränderungen komplexer sind, als Evolutionisten bisher glaubten“ (Baker 2022).
Quellen
Baker H (2022) New study provides first evidence of non-random mutations in DNA. Life Science, 14 Januar. https://www.livescience.com/non-random-dna-mutations
Borger P (2019) Artübergreifende wiederkehrende Mutationen. Stud. Integr. J. 26(2), 77-85.
Caporale LH (2003) Darwin in the Genome. The McGraw Hill Companies.
Futuyma DJ (2005) Evolutionary Biology, 3rd ed., Sinauer Associates, Sunderland, MA.
Monroe JG, Srikant T, Carbonell-Bejerano P et al. (2022) Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 602, 101–105. https://doi.org/10.1038/s41586-021-04269-6.
Spetner L (1997) Not by Chance. The Judaica Press Ltd.
Terborg P (2010) An illusion of common descent. J. Creation 24(2), 122–127.
Autor dieserNews: Peter Borger
© 2022, http://www.genesisnet.info/schoepfung_evolution/n299.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
22.04.21 Das Ausschalten von „Junk-DNA“ macht aus Stammzellen Neuronen
Ein großer Teil des Genoms codiert nicht für Proteine und enthält außerdem viele Wiederholungen. Daher wurde vielfach angenommen, dass es sich um funktionslose Sequenzen handelt. Warum aber sollte ein evolutionärer Prozess, bei dem nutzloser Ballast nur energetisch nachteilig wirkt, solche Sequenzen erhalten? Eine neue Studie zeigt nun, dass solcher vermeintlicher „Ballast“ den Zellen hilft, sich richtig zu entwickeln.
Ein großer Teil des Erbguts (Genom) besteht aus dem, was Wissenschaftler heute transponierbare und transponierte Elemente (kurz: TE) nennen, die früher als „Transposons“ oder „springende Gene“ bekannt waren. Die komplexesten TE sind endogene Retroviren (ERV) und die so genannten „long interspersed nuclear elements“ (LINE). Zusammen machen sie etwa 25% des Genoms des Menschen aus. Die meisten Biologen interpretieren diese genetischen Elemente als Überbleibsel von Virusinfektionen, die sich im Verlauf der Stammesgeschichte ereignet haben, obwohl ihnen, ähnlich wie proteincodierenden Genen, immer mehr Funktionen zugeschrieben werden. Eine wachsende Zahl Publikationen beschreibt diese Elemente als wichtige strukturelle und regulatorische Elemente des Genoms. Aktuelle Studien zeigten, dass LINE und ERV an der menschlichen Embryonalentwicklung und der interzellulären Kommunikation im Nervensystem beteiligt sind. Und überwiegend als lange nicht-codierende RNAs haben sie Aufgaben als DNA-regulatorische Elemente (Terborg 2018).
In einer kürzlich veröffentlichten Studie zeigen Forscher nun, wie eine Klasse von ERV (bekannt als HERV-K) Stammzellen veranlassen, sich in Neuronen umzuwandeln (Wang et al. 2020).
Für jede Zelle im Körper gibt es einen Zeitpunkt, an dem entschieden wird, welcher Zelltyp sie für den Rest ihres Lebens sein wird. Dies wird als Zelldifferenzierung bezeichnet. Eine von mehreren hundert unterschiedlichen differenzierten Zellen im menschlichen Körper sind Neuronen, die Zellen, aus denen das Nervensystem besteht. Forscher berichteten nun, dass die HERV-K-Gene, die früher als „Junk-DNA“ betrachtet wurden, eine sehr wichtige Rolle in diesem Prozess übernehmen (Wang et al. 2020). Sie beschreiben eine Reihe von Experimenten, die zeigten, wie einige Gene des menschlichen endogenen Retrovirus (HERV-K), die auf den Chromosomen 12 und 19 vorhanden sind, helfen können, die Differenzierung (oder Reifung) menschlicher Stammzellen in die Billionen von Neuronen zu steuern, die in unserem Nervensystem miteinander interagieren.
Die Experimente wurden von Forschern in einem Labor unter der Leitung von Avindra Nath, klinischer Direktor am NIH’s National Institute of Neurological Disorders and Stroke (NINDS), durchgeführt. Zuvor hatten Forscher im Labor von Nath gezeigt, dass die Amyotrophe Lateralsklerose (ALS), eine schwere degenerative, tödlich verlaufenden Nervenkrankheit, mit der Aktivierung von HERV-K verbunden ist. Daher ist es wichtig, die Aktivität dieser genetischen Elemente in voll ausdifferenzierten Zellen zu unterdrücken. Aber in pluripotenten Stammzellen, d. h. Zellen, die zu jedem möglichen Zelltyp werden können, ist es genau umgekehrt. In ihrer neuen Studie zeigte das Team um Nath, dass die Deaktivierung des HERV-K die Stammzellen dazu bringt, zu Neuronen zu werden.
Die Forscher führten ihre Experimente an Blutzellen durch, die sie gesunden Freiwilligen am NIH-Klinikum entnommen hatten und die sie in sog. „induzierte pluripotente Stammzellen“ verwandelten. Im Labor kann ein pluripotenter Zellzustand durch die Yamanaka-Methode induziert werden, bei der ein „Cocktail“ von vier Genen (c-Myc, Klf-4, Oct-4 und Sox-2) verwendet wird. Überraschenderweise stellten sie fest, dass die Oberflächen der Stammzellen mit hohen Mengen an HERV-K, Subtyp HML-2, einem Hüllprotein, ausgekleidet waren, das auch bestimmte Viren häufig verwenden, um sich an Zellen anzuheften und diese zu infizieren. Diese Proteine verschwanden nach und nach, als den Zellen zweimal nacheinander das erwähnte Gen-„Cocktail“ verabreicht wurde. Die erste Verabreichung versetzte die Zellen in einen Zwischenzustand, den Zustand neuronaler Stammzellen, während die zweite die Zellen dazu brachte, schließlich zu Neuronen zu werden. Die Forscher beschleunigten diesen Prozess, indem sie die HERV-K-Gene oder die HML-2-Gene in den Stammzellen ausschalteten oder sie mit neutralisierenden Antikörpern gegen das HML-2-Protein behandelten. Im Gegensatz dazu verzögerten sie die neuronale Differenzierung, indem sie die Zellen künstlich mit den HML-2-Genen anreicherten.
Schließlich entdeckte das Team, dass Interaktionen auf den Stammzelloberflächen zwischen HML-2 und einem anderen Immunzellprotein namens CD98HC die Differenzierung einschränken können, indem sie interne Signalkette auslösen, von denen bekannt ist, dass sie das Zellwachstum und die Tumorbildung kontrollieren. Für die Zukunft plant das Team zu erforschen, wie HERV-K-Gene die Verdrahtung eines Nervensystems gestalten können. Wir können weitere unerwartete Funktionen für diese Elemente erwarten, die sich als Regulatoren der Zelldifferenzierung und -reifung erweisen. Dass sie die Überreste uralter Viren sind, ist evolutionäres „Storytelling“. Vielmehr könnten die heutigen Viren ihren Ursprung in diesen genetischen Elementen haben. Diese Forschungen geben auch Einblicke in die fein abgestimmte Wechselwirkung von Genen bei der Differenzierung und lassen erahnen, wie welches Potenzial an Fehlfunktionen denkbar ist; ein Wunder, dass die Entwicklung typischerweise korrekt verläuft!
Literatur
Terborg P (2018) ERVs and LINEs – along novel lines of thinking. J. Creation 32, 8–11.
Wang T et al. (2020) Regulation of stem cell function and neuronal differentiation by HERV-K via mTOR pathway. Proc. Natl. Acad. Sci. 117, 17842–17853.
Autor dieserNews: Peter Borger
© 2021, http://www.genesisnet.info/schoepfung_evolution/n290.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
22.01.21 Seeanemonen: Evolution oder Abruf eines Programms?
Das Ausschalten eines Regulationsgens der Seeanemone Nematostella vectensis führt dazu, dass ein neuer Zelltyp ausgebildet wird, eine Art Klebzelle, die bei dieser Art bisher nicht beobachtet wurde. Wissenschaftler diskutieren diesen Befund im Rahmen von Evolution; er verweist aber viel klarer auf die Existenz bereits angelegter Strukturen. Aus der Sicht einer Schöpfung kann das so verstanden werden, dass Lebewesen mit mehr abrufbaren Fähigkeiten ausgestattet sind, als sie gewöhnlich benötigen.

Abb. 1: Nematostella vectensis in einer Petrischale. (Wikimedia: Cymothoa exigua, CC BY-SA 3.0)
Lebewesen besitzen schlummernde Bauplanmodule, die im Normalfall nicht genutzt werden, aber unter bestimmten Bedingungen abrufbar sind. Wer hätte beispielsweise gedacht, dass manche krautige Pflanzen eine Art „Holzmodul“ besitzen, das sie unter bestimmen Bedingungen ausprägen können und dadurch verholzen? (Losos 2018, 93). Hat dies etwas mit Evolution zu tun? Wohl kaum, denn ein Bauelement, das durch einen Umweltreiz oder eine geringfügige genetische Änderung fix und fertig abgerufen werden kann, war offenbar schon vorher da; seine Entstehungsweise ist davon unberührt.
Über ein erstaunliches Beispiel eines latent (im Verborgenen) vorhandenen Organs bei Quallen berichtete kürzlich Leslie Babonis von der Cornell University auf der virtuellen Jahrestagung der Society for Integrative and Comparative Biology (Pennisi 2021). Quallen sind berüchtigt wegen ihrer Nesselzellen, deren Stich Hautreaktionen und heftige Schmerzen verursachen. Babonis hat herausgefunden, dass ein einziger genetischer Schalter stechende Nesselzellen in Zellen umwandeln kann, die an einer Unterlage kleben können. Dies ermöglicht dem Tier, im Verlauf der Entwicklung von der frei beweglichen Larve zum festsitzenden (sessilen) Organismus neue Oberflächen zu besiedeln.
Die Nesselzellen der Quallen haben mehrere Erscheinungsformen. Die Stachelzellen (Nematozyten) schießen winzige, giftbeladene Harpunen ab, während einige Nesselzellen in Seeanemonen und Korallen Fäden aus klebrigem Material absondern, mit denen sich die Tiere in Substraten wie Schlamm verankern können.
Babonis untersuchte mit ihrem Team die 5 cm lange, durchsichtige Seeanemone Nematostella vectensis. Die Forscher schalteten das Sox2-Gen aus, das bei vielen anderen Tieren für die neurale Entwicklung wichtig ist. Es zeigt sich, dass als Folge davon Nematozyten in den Tentakelspitzen krumme Harpunen ausbildeten, Nematozyten aber an der Körperwand fehlten; an ihrer Stelle wurden fette Nesselzellen, sogenannte robuste Spirozyten, ausgebildet, die (bei anderen Arten) für ihre Klebrigkeit bekannt sind. Diese Zellen waren bei dieser Art bislang noch nie beobachtet worden. Offenbar hat das Sox2-Gen die Funktion eines Schalters. Wenn es aktiv ist, bilden sich Stachelzellen, andernfalls bilden sich Spirozyten, aber nur in der Körperwand. Ob Sox2 auch bei anderen Nesseltieren für die Spirozytenbildung wichtig ist, ist bisher nicht bekannt.
Solche Regulationsgene, die wie Schalter wirken, sind an sich keine neue Entdeckung; man kennt sie als homöotische Mutationen. Zum Beispiel können durch solche Mutationen die Antennen eines Insektes in Beine umgewandelt werden. Ein neuer Befund ist jedoch, dass solche Änderungen auch bei als einfach geltenden und stammesgeschichtlich an der Basis stehenden Formen vorkommen. „Man erwartet normalerweise nicht, dass Organismen in der Lage sind, Organe zu produzieren, die sie normalerweise nicht entwickeln“, kommentiert Nicole Webster, Evolutionsbiologin an der Clark University (Pennisi 2021). Man kann das in der Tat nicht erwarten, wenn es sich um eine Neuentstehung handeln würde. Aber das ist offenkundig nicht der Fall. Das plötzliche Auftreten der Klebzellen (Spirozyten) ist vielmehr ein eindrucksvolles Beispiel dafür, dass Lebewesen Bauplanmodule gleichsam in petto haben, die bereits fertig ausgebildet sind und auf Abruf bereit stehen. Dabei spielt es nur eine untergeordnete Bedeutung, ob es sich bei den experimentell umgewandelten Nesselzellen um echte Spirozyten handelt. Die Information zur Ausbildung dieser spezialisierten Zellen war in jedem Fall bereits vorhanden.
Evolution? Außer dem beschriebenen Phänomen ist auch bemerkenswert, wie dieser Befund von den Wissenschaftlern kommentiert wird: Er wird in den Kontext von Evolution gestellt, obwohl überhaupt keine evolutive Veränderung beobachtet wurde. So meint Nicole Webster, die Studie zeige, wie die Evolution neuartiger Eigenschaften auf einer kleinen Skala – verschiedene Zelltypen – erfolge, und dabei zu größeren Konsequenzen führen könne. Pennisi kommentiert: „Die Arbeit legt nahe, dass diese Art von Schalter dazu beigetragen haben könnte, evolutionäre Veränderungen über Hunderte von Millionen Jahren voranzutreiben.“ Sie zitiert Billie Swalla, Evo-Devo-Forscherin an der University of Washington in Seattle: „Die Nesselzellen sind ein großartiges Modellsystem, um die Evolution und Diversifizierung neuartiger Zelltypen zu untersuchen.“ Und Kristen Koenig, Evolutionsbiologin an der Harvard University, sieht es so: „Die Entdeckung [liefert] aussagekräftige Beobachtungen darüber, wie sich neue Zelltypen entwickeln und an neuartigen Orten ankommen.“
Hier wird offensichtlich Evolution in den experimentellen Befund hineingelesen. Man hat den Eindruck, dass hier Befunde reflexartig nach einem evolutionären Prinzip eingeordnet werden. Wirkliche Erklärungen werden damit aber nicht geliefert und es stellen sich stattdessen neue Fragen: Wie sind die spezialisierten Typen der Nesselzellen ursprünglich entstanden? Wie konnte die Fähigkeit, Klebzellen zu bilden, in einem latenten Zustand erhalten bleiben? Aus der Sicht einer Schöpfung können die Befunde dagegen zwanglos verstanden werden: Lebewesen sind mit mehr Fähigkeiten ausgestattet, als sie gewöhnlich benötigen, können diese aber unter bestimmten Umständen abrufen. Das ist ein Indiz für vorausschauendes Handeln und mithin für einen Schöpfer.
Quellen
Losos JB (2018) Glücksfall Mensch. Ist Evolution vorhersehbar? München.
Pennisi E (2021) Anemone shows mechanism of rapid evolution. Science 371, 221.
Autor dieserNews: Reinhard Junker
© 2021, http://www.genesisnet.info/schoepfung_evolution/n285.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
17.04.20 Proteom-basierter Stammbaum mit interessanten Resultaten
Stammbäume prägen evolutionäres Denken nachhaltig. Ein neues Modell zur Erstellung von Stammbäumen liefert eine Baumstruktur, bei der sich die Bereiche der Mikroorganismen, Pilze, Pflanzen und Tiere bereits sehr früh und in einem vergleichsweise kurzen Zeitraum aufspalten.
Song-Hou Kim, ein in Berkeley, CA (USA) forschender Biophysiker, hatte 2009 eine Methode zum Vergleich von Sequenzdaten ganzer Genome (Nukleotidabfolgen des kompletten Erbguts) veröffentlicht, bei der auf die Ausrichtung der Sequenzen vor dem Vergleich (Alignment) verzichtet wird (Sim et al. 2009). Im Alignment werden Sequenzen entsprechend zugrundgelegter Modelle so angeordnet, dass sie maximale Ähnlichkeit aufweisen bzw. die Passung der Abfolgen bei verschiedenen Genomen maximal ist. In der von Sim et al. etablierten Methode wird dagegen die Häufigkeit bestimmter Muster in den Sequenzen zum Vergleich genutzt. Dies erlaubt auch Sequenzen ungleicher Länge miteinander zu vergleichen; diese müssen auch keine auffällige Ähnlichkeit aufweisen.
Die Arbeitsgruppe von Kim hat inzwischen die Leistungsfähigkeit dieser Methode an zellkernlosen Einzellern, also Prokaryonten (Jun et al. 2010) und an Pilzen (Choi et al. 2017) demonstriert. Jetzt haben Choi & Kim (2020) anhand des Proteoms von 4.023 Organismen einen Stammbaum des Lebens errechnet. Als Proteom wird die Gesamtheit aller im Genom codierter Proteine bezeichnet. Die Proteinsequenzen stammen aus entsprechenden Datenbanken (vor allem vom National Center for Biotechnology Information, NCBI).
Die Einführung der Autoren in ihrer Veröffentlichung ist erfreulich nüchtern, wenn sie schreiben: „Der Stammbaum des Lebens von Organismen ist ein konzeptioneller und bildhafter Baum, in dem eine einfache Erzählung des evolutionären Verlaufs und der Verwandtschaft unter den heutigen Lebewesen dargestellt wird. Ein solcher Baum kann nicht experimentell bestätigt werden, sondern wird aus Eigenschaften der Organismen rekonstruiert.“1 Damit wird zum Ausdruck gebracht, dass ein Ähnlichkeitsbaum nicht mit einem Abstammungsbaum gleichzusetzen ist, sondern dass Letzterer eine Interpretation ist.
Der von Choi et al. (2020) präsentierte Stammbaum des Lebens auf der Basis des Proteoms unterscheidet sich in seiner Grundstruktur von den typischen Bäumen, die auf der Basis einzelner Gene errechnet werden (s. Abb. 1). Die Unterschiede betreffen sowohl die Gruppierungen als auch deren Verknüpfung. Die Knoten (Verzweigungspunkte) haben aufgrund der Besonderheit der neu entwickelten Methode eine andere Bedeutung als in den etablierten Stammbaumdarstellungen. Der Knoten repräsentiert nicht einen hypothetisch gemeinsamen Vorfahren aller im weiteren Astverlauf dargestellten Organismen, wie das in üblichen Stammbäumen interpretiert wird. Die Knoten in dem von Choi & Kim (2020) präsentierten Stammbaum sind nach den Worten der Autoren eine Gruppe von Gründungsvorläufern mit stark unterschiedlichem Proteom; als bildhaften Vergleich führen sie einen mit Mosaiksteinen gefüllten Beutel an.
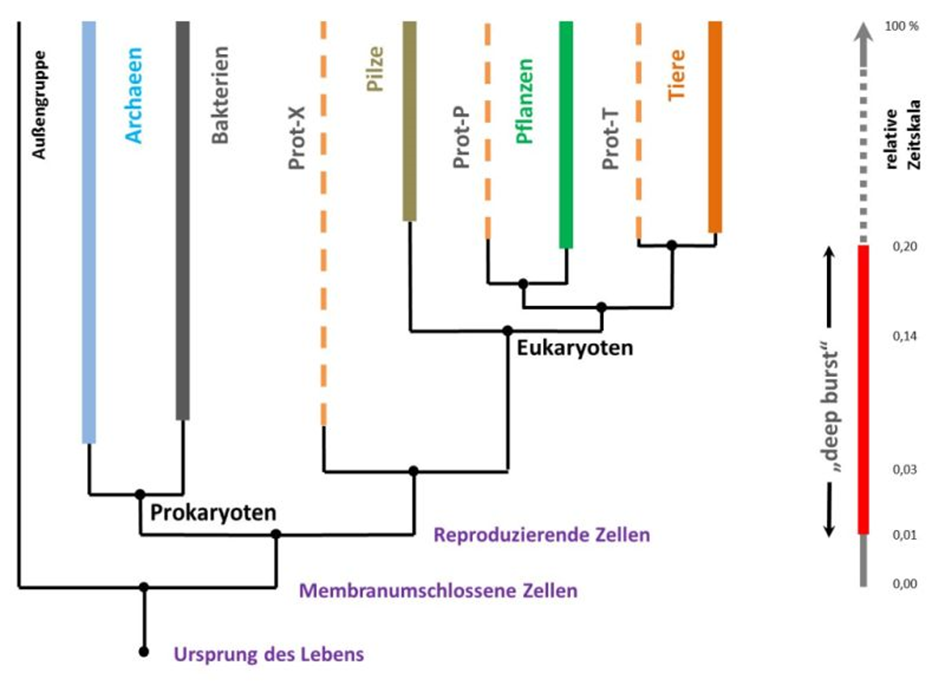
Abb. 1: Vereinfachter Stammbaum der Lebewesen auf Basis des Proteoms. Die fünf Großgruppen Archaeen, Bakterien (Prokaryoten), Pilze, Pflanzen und Tiere (Eukaryoten) sind bereits sehr früh in einem als „deep burst“ bezeichnete kurzen Zeitraum aufgezweigt. Man beachte die relative Skala: Nach 0,2% der Geschichte der Lebewesen sind alle Großgruppen getrennt. Dier Stamm hat weniger als ein Tausendstel der Länge des Baumes. Die Außengruppe ist eine für den Rechenprozess notwendige zufällig gemischte Proteinsequenz. Bei Prot-X,-P und –T handelt es sich um Gruppen von Protisten, die nicht einzuordnen waren und den Eukaryoten (Prot-X), Pflanzen (Prot-P) bzw. den Tieren (Prot-T) gegenüberstehen. (Nach Choi & Kim 2020, Abb. 2)
In dem von Choi & Kim (2020) errechneten Stammbaum des Lebens wird der Abstand der Knoten, also der Verzweigungen der Baum-Äste durch die aufsummierten genomischen Informationsdifferenzen bestimmt. Der so errechnete Baum weist sehr tief im Baum, also nahe dem Wurzelbereich, fünf Hauptgruppen, die in der systematischen Biologie den Reichen entsprechen. Das heißt, der Stammbaum weist keinen nennenswerten Stamm auf. Zuerst spalten sich die Prokarya (die Autoren bezeichnen sie auch als Akarya) und Eukarya auf, die dann jeweils in Archaea und Bakteria bzw. Pilze, Pflanzen und Tiere aufspalten. Dies verleiht dem Baum eine Form, in der ein sehr kurzer Stamm sehr schnell buschförmig breit wird. Die Autoren bezeichnen das als „deep burst“, also eine Explosion, die in einem sehr frühen Stadium lokalisiert ist.
Das Modell von Choi & Kim (2020) führt also zu dem Schluss, dass die spezifischen Eigenheiten der Proteome von Archaeen, Bakterien, Pilzen, Pflanzen und Tieren sehr früh und in einem vergleichsweise kurzen Zeitraum auftauchen.
Quellen
Choi J & Kim S-H (2017) A genome Tree of Life for the Fungi kingdom. Proc. Natl. Acad. Sci. USA 114, 9391–9396.
Choi J & Kim S-H (2020) Whole-proteom tree of life suggest a deep burst of organism diversity. Proc. Nat. Acad. Sci USA, doi: 10.1073/pnas.1915766117.
Jun S-R, Sims GE, Wu GA & Kim S-H (2010) Whole-proteome phylogeny of prokaryotes by feature frequency profiles: An alignment-free method with optimal feature resolution. Proc. Natl. Acad. Sci. USA 107, 133–138.
Sim GE, Jun S-R, Wu GA, Kim S-H (2009) Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proc. Nat. Acad. Sci. USA 106, 2677-2682.
Anmerkung
1 An organism tree of life (organism ToL) is a conceptual and metaphorical tree to capture a simplified narrative of the evolutionary course and Kinship among the extant organisms. Such a tree cannot be experimentally validated but may be reconstructed based on characteristics associated with the organisms.
Autor dieserNews: Harald Binder
© 2020, http://www.genesisnet.info/schoepfung_evolution/n279.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
27.03.20 Molekularbiologie des Coronavirus und die Coronakrise
Das derzeit zirkulierende und die Krankheit COVID-19 auslösende Coronavirus wurde von einem internationalen Konsortium von Virusexperten auf den Namen SARS-CoV2 getauft. Es wird so genannt, weil es dem Corona-Virus ähnelt, das im Jahr 2003 SARS verbreitete und als SARS-CoV bekannt ist. Die Genetik des COVID-19-Virus zeigt, dass es sich sehr wahrscheinlich um eine Variante des alten SARS-CoV1-Virus aus dem Jahr 2003 handelt. COVID-19 ist also eigentlich SARS.
Coronaviren sind bekannte Atemweg-Viren. Der Name Corona (lat.: Kranz, Krone) leitet sich von den Proteinen ab, die auf der Virushülle herausragen und dem Virus, durch das Elektronenmikroskop gesehen, eine Art Krone verleihen. Coronaviren sind so genannte „positive sense, single stranded RNA-Viren“. Das bedeutet, dass sie anstelle von DNA ein einsträngiges RNA-Molekül als genetisches Material besitzen. Wo Coronaviren ihren Ursprung haben, ist derzeit nicht geklärt, allerdings weist ihre genetische Ausstattung darauf hin, dass sie aus dem Genom (= Erbgut) von Wirbeltieren stammen. Durch Rekombination im Wirtsgenom wird manchmal genetisches Material in ihr Genom eingefügt oder geht verloren. So enstehen neue Varianten. Gegenwärtig kennen wir sieben Typen von Coronaviren, die den Menschen infizieren können. Nach einer Infektion entwickelt der Mensch krankhafte Atemwegssymptome unterschiedlichen Schweregrades. Zwei der Coronaviren, HCoV-229E und HCoV-OC43, verursachen Erkältungen. Andere Coronaviren können zu schwereren Infektionen der Atemwege führen und potenziell tödlich sein.
SARS-CoV war das erste Coronavirus, das im Jahr 2003 zu einer Bedrohung wurde. Der Kongress der Lungenspezialisten, den ich damals in Australien besuchte, widmete dem Coronavirus SARS-CoV, das damals das schwere respiratorische Syndrom (SARS) verursachte, besondere Aufmerksamkeit. Dies lag daran, dass SARS eine Sterblichkeitsrate von 9 % hatte und als ernsthafte Gefahr für die westliche Gesellschaft angesehen wurde. Glücklicherweise war SARS-CoV nicht so ansteckend wie befürchtet. Innerhalb weniger Monate war das Virus verschwunden. Doch die Virologen wussten, dass dies nicht das letzte tödliche Coronavirus sein würde. Im Jahr 2012 tauchte der nächste Coronavirus, MERS-CoV, auf. Mit einer Sterblichkeitsrate von über 30% der Infizierten ist es das bisher aggressivste Coronavirus. Auch dieses Virus verschwand vom Radar, ohne eine Pandemie auszulösen. Dennoch rechneten Virologen mit einen weiteren Coronavirus. Jetzt ist es leider da: SARS-CoV2. Dieses Virus verursacht COVID-19. Und es hat bereits eine weltweite Epidemie (Pandemie) ausgelöst. Mit einer Sterblichkeitsrate von wahrscheinlich unter 1% ist SARS-CoV2 nicht so gefährlich wie frühere Corona-Viren, aber es ist weitaus ansteckender (höhere Infektiosität).[1]
Alle aggresiven Coronaviren haben sehr ähnliche molekularbiologische Strukturen. Das virale Genom kodiert mehrere Proteine mit einzigartigen Funktionen, darunter ein S- und ein N-Protein. Das N-Protein wird derzeit als diagnostischer Marker verwendet. Das heißt, wenn dieses Protein in Patienten nachgewiesen wird, handelt es sich um eine aggressive, d.h. hochinfektiöse Form der Corona. Das S-Protein bildet die Krone des Virus. Es hat eine Domäne (= Bereich eines Proteins), die sich an ein Rezeptorprotein auf der menschlichen Zelle (ACE2-Rezeptor) anlagert. ACE2-Rezeptoren befinden sich auf den Lungenzellen und regulieren den Blutdruck. Experimente mit Labortieren bestätigten, dass das Virus den ACE2-Rezeptor nutzt, um in die Zellen einzudringen. Mäuse, denen dieser Rezeptor fehlt, sind gegen das SARS-CoV-Virus resistent und entwickeln keine SARS-Symptome.[2] Das S-Protein stellt das attraktivste Ziel für die Entwicklung von Impfstoffen und Antikörpern dar, da die Protease-Aktivität des S-Proteins es ermöglicht, in die Zellen des menschlichen Körpers einzudringen. (Proteasen sind Enzyme, die Peptidbindungen von Proteinen spalten.) SARS-CoV2 unterscheidet sich genetisch von SARS-CoV durch vier kleine Insertionen (Einfügungen) im S-Protein.
Das jetzt zirkulierende Coronavirus ist sehr eng mit dem SARS-CoV-Virus von 2003 verwandt. Eine Studie von März 2020 belegt, dass das genetische Material des SARS-CoV2 Virus zu 96,11% dem SARS-Virusstamm RaTG13 entspricht.[3] Würde man jedoch die gleiche Methode anwenden, wie sie Evolutionsbiologen beim Vergleich der Gene von Mensch und Schimpanse anwenden (d.h. die Insertionen werden nicht als Unterschiede gezählt) wären die Genome beider Viren zu mehr als 99 Prozent gleich.
Die Gruppe um den Virologen Markus Hoffman, der am Leibniz-Institut für Primatenforschung in Göttingen forscht, hat jüngst gezeigt, dass SARS-CoV2 denselben ACE2-Rezeptor zur Infektion menschlicher Zellen verwendet wie SARS-CoV.[4] Diese Studien legen ebenfalls nahe, dass das Coronavirus, das COVID-19 verursacht, sehr eng mit dem SARS-CoV-Virus von 2003 verwandt ist. Das COVID19-Virus hat die gleichen Gene wie das SARS-Virus von 2003 und es gelangt auf die gleiche Weise in die menschliche Zelle, d.h. über den ACE2-Rezeptor.
Im Laufe der Zeit werden Viren durch Mutationen in ihrer Wirkung typischerweise immer schwächer, nicht aggressiver, wie man meinen könnte. RNA-Viren mutieren sehr schnell und die beständige Anhäufung von Mutationen macht RNA-Viren immer weniger gefährlich, gleichzeitig können sie aber infektiöser werden. Die Virusvarianten, die die für Menschen hochpathogen und tödlich sind, werden weniger. Am Ende ist ein Arrangement, eine Art „friedliches Zusammenleben“ mit ihren Wirten zu erwarten. So war beispielsweise die H1N1-Schweinegrippe 2009 am stärksten in der ersten Pandemie im Sommer 2009. Im Winter 2010/2011 hatte sie sich beruhigt und verhielt sich eher wie eine typische saisonale Grippe. Durch die Insertionen im S-Protein ist das jetzt sich ausbreitende SARS-CoV2 Virus zwar infektiöser als SARS-CoV aus 2003, glücklicherweise auch weniger gefährlich – genau so, wie man es von einem alternden mutierten Virus erwarten würde.
Zwischen dem Ausbruch von SARS im Jahr 2003 und dem Ausbruch von COVID-19 im Jahr 2019 liegen 16 Jahre. Wurde in diesen 16 Jahren eine Behandlung für SARS entwickelt? Wurde ein Impfstoff entwickelt? Wurde ein blockierender Antikörper entwickelt? Alles Wissen dazu war vorhanden. Und man wusste, dass mit einem weiteren Ausbruch von Coronaviren zu rechnen war, höchstwahrscheinlich in China. Die schockierende Tatsache ist, dass die entscheidenden Anstrengungen zur Bekämpfung des nächsten Ausbruchs eines tödlichen Coronavirus unterlassen wurden.
Es gab reichlich Gelegenheit, Heilmittel und Impfstoffe zu entwickeln, um die Corona-Krise zu verhindern, die de facto vor 16 Jahren begann. Hätte man auf die Corona-Experten gehört, könnten wir möglicherweise über eine entsprechende Therapie verfügen und die aktuelle Coronakrise wäre weniger gravierend. Mit konsequenteren Entscheidungen im vergangenen Jahrzehnt hätte SARS-CoV2 vermutlich bereits in Asien gestoppt werden können. Mit einem aus verschiedenen Gründen veranlassten Stopp der Impfstoffentwicklung wurde die aktuelle Corona-Krise begünstigt. Hier wird besonders deutlich erkennbar, wie weitreichend wirtschaftspolitische Entscheidungen sein können.
Quellen
[1] Zur Sterblichkeitsrate bei Ansteckungen mit SARS-CoV2 scheint es noch keine gesicherten Erkenntnisse zu geben. Es werden Angaben von 0,37% bis 3-4% gemacht. Der Virologe Hendrik Streeck ermittelte auf der Basis einer repräsentativen Stichprobe eine Sterblichkeitsrate von 0,37% (https://www.faz.net/aktuell/gesellschaft/gesundheit/coronavirus/corona-in-heinsberg-virologe-streeck-sieht-moegliche-lockerung-16718884.html). Dieser Wert könnte immer noch zu hoch sein, da nur durch Obduktion die genaue Todesursache festgestellt werden kann; das wird aber in der Regel nicht gemacht.
[2] Kuba K et al. (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nature Medicine 11, 875-879. https://www.ncbi.nlm.nih.gov/pubmed/16007097
[3] Yu H et al. (2020) Genomic analysis of a 2019-nCoV strain in the first COVID-19 patient found in Hangzhou, Zhejiang, China. Zhonghua Yu Fang Yi Xue Za Zhi. 2020 Mar 15;54(0):E026. doi: 10.3760/cma.j.cn112150-20200217-00128. https://www.ncbi.nlm.nih.gov/pubmed/32171191
[4] Hoffmann M et al. (2020) SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell, 2020 Mar 4. pii: S0092-8674(20)30229-4. doi: 10.1016/j.cell.2020.02.052. [Epub ahead of print] https://www.ncbi.nlm.nih.gov/pubmed/32142651
Autor dieserNews: Peter Borger
© 2020, http://www.genesisnet.info/schoepfung_evolution/n277.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
09.03.20 Supergene ermöglichen eine sehr schnelle Veränderung
Das Beispiel des Sexualpolymorphismus beim Kampfläufer
Biologische Variation beruht nicht nur auf langsamer und allmählicher Anhäufung von genetischen Mutationen, wie es dem Neodarwinismus entspricht, sondern kann auch durch große DNA-Blöcke – sogenannte Supergene – moduliert werden. Dabei sind eine oder wenige genetische Veränderungen erforderlich, damit neue Phänotypen ausgeprägt werden können.
Im Jahr 2017 traf sich eine internationale Gruppe von Evolutionsbiologen in Groningen, Niederlande, um die neuesten Erkenntnisse auf ihrem Gebiet zu besprechen. Zu den Organismen, über die sie berichteten, gehörten Kampfläufer, Honigbienen und Zebrafische. Eine große Überraschung, die auf diesem Kongress präsentiert wurde, war der Befund, dass diese Organismen alle über einen eingebauten Mechanismus – die Wissenschaftszeitschrift Science nannte es eine „Geheimwaffe“ – verfügen, um ihre „Evolution“ zu lenken (Pennisi 2017). Die Biologen beschrieben in allen Fällen die gleichen genetischen Module, sog. Supergene, die es den Organismen ermöglichen, blitzschnell neue Eigenschaften abzurufen.
Kragen und Haube
Kampfläufer sind etwa 30 cm große Wiesenvögel. Wie der Kiebitz und die Uferschnepfe werden sie zu den Watvögeln gezählt und sind wie diese in den letzten Jahrzehnten immer seltener geworden. Wer schon einmal eine Gruppe von Kampfläufern gesehen hat, dem ist sicher aufgefallen, dass es große Unterschiede zwischen männlichen und weiblichen Tieren gibt. Biologen nennen dieses Phänomen „Sexualdimorphismus“. Männliche Kampfläufer haben einen auffälligen Federkragen und eine Haube, die sie von Zeit zu Zeit eindrucksvoll aufrichten. Bei den Weibchen fehlen Kragen und Haube vollständig.
Fast ebenso auffällig sind die großen Unterschiede zwischen den Kragen verschiedener Männchen. Es gibt zwei auffallend unterschiedliche Varianten bei den Männchen, die vor allem die Farbe ihrer Kragen betreffen. Einige Männchen besitzen einen weißen Kragen, der sich deutlich vom grünen Gras abhebt. Die Mehrheit der Männchen hat jedoch einen rötlich-braunen Kragen. Darüber hinaus gibt es männliche Kampfläufer, die den Weibchen sehr ähnlich sehen, weil sie keinen Kragen und keine Haube haben. Biologen haben diese weiblich aussehenden Männchen erst vor einigen Jahrzehnten entdeckt. Die Männchen mit Kragen konkurrieren um die Gunst der Weibchen, während die Männchen ohne Kragen ohne Auseinandersetzung auskommen. Da sie den Weibchen sehr ähnlich sind, können sie sich heimlich anschleichen und sich ungehindert – ohne Rituale der Kämpfer – mit den Hennen paaren. Diese drei klar definierten männlichen Varianten kommen in allen Populationen in ungefähr gleichen Anteilen vor. Die Variation bei den Männchen scheint also erblich bedingt zu sein. Bis vor kurzem war der Ursprung dieser Variation unbekannt, aber dank der Molekularbiologie weiß man jetzt, wie sie entsteht.
Das Super-Gen
Das Erbgut der Kampfläufer enthält eine enorme Menge an Information, die die Merkmale des Vogels betreffen. Diese Information ist auf der DNA der Chromosomen gespeichert. Biologen haben herausgefunden, dass die Ausrichtung dieser Information auf den Chromosomen von großer Bedeutung zu sein scheint. Es stellte sich nämlich heraus, dass bei den Männchen ohne Kragen ein großes Stück DNA in umgekehrter Reihenfolge auf Chromosom 11 angeordnet ist. Dabei handelt es sich um einen DNA-Abschnitt mit mehr als 4 Millionen Nukleotiden (DNA-Buchstaben), der Dutzende von Genen enthält und als eine einzige funktionelle Einheit an die Nachkommen weitergegeben wird. Ein solches genetisches Modul wird als Supergen bezeichnet. Dieses Supergen trägt die Information für die typischen männlichen Merkmale der Kampfläufer, einschließlich des farbigen Kragens. Wenn das Supergen umgekehrt angeordnet ist, verschwinden diese äußeren Merkmale, aber die Information selbst geht nicht verloren. Bei den kragenlosen Männchen wurden die typischen äußeren Merkmale also durch eine Chromosomeninversion abgeschaltet.
Diese Veränderungen („Evolution“) beim Kampfläufer werden am besten als Positionseffekte der bereits vorhandenen genetischen Information interpretiert. Positionseffekte sind oft epigenetischer Natur und lassen sich leicht als veränderte Ausprägung einer bereits vorhandenen genetischen Information erklären. In diesem speziellen Fall wird die Ablesung des Supergens sogar vollständig unterdrückt, was dazu führt, dass die Männchen wie Weibchen aussehen. Diese Veränderung findet ohne die Notwendigkeit neuer genetischer Information statt. Der Prozess erfordert keine Millionen von Jahren der Selektion, da er auf einer einzigen genetischen Veränderung, nämlich der Inversion des Supergens, beruht. Es handelt sich also um eine sofort eintretende Änderung, die unmittelbar stattfindet, mit direkten Folgen für die Nachkommenschaft.
Überall Supergene
Wenn man die genetischen Grundlagen für bestimmte Merkmale genauer untersucht, findet man in vielen Fällen Supergene. Im Erbgut von Honigbienen, Singvögeln und Zebrafischen werden ähnliche modulartige Supergene zur Erzeugung von Variation verwendet. Immer wieder kann man feststellen, dass ein oder mehrere DNA-Abschnitte umgekehrt im Erbgut ausgerichtet sind. Es ist offenbar ein allgemeines genetisches Prinzip, das es ermöglicht, neue Varianten und Arten auf einfache, aber superschnelle Weise hervorzubringen, ohne dass neue genetische Information benötigt wird. Solche Veränderungen dauern also nicht Millionen von Jahren.
In der Vergangenheit wurde diese Art des schnellen Wandels von manchen Biologen vorhergesagt. Bemerkenswerterweise nahmen diese Biologen nicht den Darwinismus, sondern biologische Beobachtungen zum Ausgangspunkt. Der Zellbiologe John A. Davison (1930-2012) fasste diese Beobachtungen unter der Bezeichnung „vorgeschriebene Evolutionshypothese“ („prescribed evolutionary hypothesis“) zusammen, und Todd und Kolnicky entwickelten die „Karyotyp-Spaltungstheorie“ („karyotype fission theory“) (Borger 2018). Nach beiden Theorien entstehen neue Eigenschaften durch eine Neuanordnung des Erbguts, d. h. durch Positionseffekte. Außerdem wird die neue Ausrichtung eine völlig andere epigenetische Steuerung der Gene bewirken.
Die Genetik der Lebewesen erscheint zum Teil wie ein vorgefertigter Baukasten, wie Legosteine. Mit einer begrenzten Anzahl von Bausteinen kann man viele verschiedene Formen realisieren. Auf die gleiche Weise kann man mit einer begrenzten Anzahl von genetischen Modulen eine enorme Menge an Variation erzeugen. Die Molekularbiologie zeigt einmal mehr, wie schön und wunderbar das Leben geschaffen worden ist.
Quellen
Pennisi E (2017) ‘Supergenes’ drive evolution. Science 357, 1083. http://science.sciencemag.org/content/357/6356/1083.full
Borger P (2018) Darwin Revisted. Scholars Press. https://www.amazon.de/Darwin-Revisited-understand-biology-century/dp/6202315113
Der Autor veröffentlichte einen ähnlichen Artikel auf Holländisch für Weet (Weet 2018; 49:22)
Autor dieserNews: Peter Borger
© 2020, http://www.genesisnet.info/schoepfung_evolution/n276.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
08.10.18 Nachahmung von Evolution oder intelligentes Design?
Nobelpreis 2018 für Chemie sorgt für irreführende Schlagzeilen
Der diesjährige Chemie-Nobelpreis wurde an drei Forscher vergeben, die bahnbrechende Erfolge mit „gerichteter Evolution“ erreicht haben. Der Begriff „gerichtete Evolution“ ist eigentlich ein Widerspruch in sich, da natürliche Evolution ungerichtet verläuft, wie von Evolutionstheoretikern selbst immer wieder betont wird. Auch wenn die (Wissenschafts-)Presse die Vergabe des Nobelpreises mit „Nachahmung von Evolution“ kommentiert, kann dies nicht darüber hinwegtäuschen, dass die Wissenschaftler nur in einem eingeschränkten Sinne den evolutiven Mutations-Selektions-Mechanismus imitiert haben. Ihre Erfolge beruhen vor allem auf einer intelligenten, zielorientierten Herangehensweise. Der Vergleich mit natürlicher Evolution ist daher irreführend.
„Der diesjährige Chemie-Nobelpreis geht an drei Forscher, die die Prinzipien der biologischen Evolution für die Entwicklung neuer Enzyme und Antikörper nutzbar gemacht haben.“1 So oder ähnlich wurde am 3. Oktober die Vergabe des Chemie-Nobelpreises an drei Forscher gemeldet. Frances H. Arnold, George P. Smith und Sir Gregory P. Winter haben Methoden entwickelt, die als „gerichtete Evolution“ bezeichnet werden. Manche Pressemeldungen dazu erweckten den Eindruck, als könnten funktionale Proteine von alleine durch natürliche Prozesse entstehen und die Forscher hätten diese Prozesse nur nachgeahmt und beschleunigt.2
Wie funktioniert „gerichtete Evolution“? Es geht darum, Proteine mit gewünschten Eigenschaften herzustellen bzw. zu optimieren (z. B. in Waschmitteln, in der Lebensmittelherstellung, in der Textilindustrie oder in der Tierernährung). Dazu werden bereits vorhandene, funktionale Proteine herangezogen und künstlich in großer Zahl mutiert und die gewünschten Mutanten ausgelesen. Die frisch gebackene Nobelpreisträgerin Frances Arnold ging dabei wie folgt vor: Sie isolierte ein proteinkodierendes Gen und produzierte eine große Menge an Mutanten in einem Reagenzglas mithilfe eines Enzyms, das das Gen kopiert. Für diesen Kopiervorgang wird durch Wahl des Enzyms und des Versuchsablaufs der ganze Prozess so angelegt, dass durch Kopierfehler Varianten des Gens entstehen. Die mutierten Genkopien werden in Bakterien integriert, welche die entsprechenden Proteinvarianten produzieren. Auf diese ausgeklügelte Weise werden die Mutationsrate und damit die Vielfalt der Proteinvarianten stark erhöht. Anschließend untersuchte die Forscherin die Funktion der so erzeugten verschiedenen Proteine. Diejenigen Varianten, die die gewünschte Funktion am besten erfüllten, wurden selektiert (ähnlich wie Evolution durch Selektion der Bestangepassten).
Dieser Prozess der Erzeugung von Varianten mit nachfolgender Selektion auf die gewünschte Funktion wurde mehrfach mit den jeweils am besten funktionierenden Genen des vorhergehenden Zyklus wiederholt. Die beste Proteinvariante wurde also ausgewählt und das entsprechende variierte Gen aus den Bakterien isoliert. Dann wurden mit dieser Genvariante mit derselben Prozedur erneut Mutationen in einem Reagenzglas erzeugt und eine große Menge an Mutanten der zweiten Generation produziert. Diese neuen Mutanten wurden wieder in Bakterien platziert, diese produzierten die neuen Proteinvarianten, die besten wurden wieder ausgelesen usw. Auf diese Weise wurden im Laufe vieler Generationen spezialisierte Proteine erzeugt. In manchen Fällen gelang es sogar, Proteine mit einer neuen Funktion zu erzeugen, wobei aber auch in diesen Fällen ein bereits existierendes Gen bzw. das entsprechende Genprodukt, das Protein, optimiert wurde.
Der Mutations- und Ausleseprozess ist es, der mit gewissen Einschränkungen mit natürlicher Evolution verglichen werden kann. Allerdings gibt es erhebliche Unterschiede. Die beiden wichtigsten sind: 1. Die Nachahmung der Evolution startet mit natürlich vorkommenden, bereits funktionalen Proteinen; diese werden nicht durch evolutive oder Evolutions-analoge Prozesse erzeugt, sondern aus Lebewesen entnommen. 2. Die Auslese erfolgt auf ein vorgegebenes Ziel hin. Genau das kann aber in der natürlichen Evolution nicht vorausgesetzt werden und wird dort sogar ausdrücklich bestritten. Bei der „gerichteten Evolution“ ist also anders als bei natürlicher Evolution Planung entscheidend im Spiel.
Ein weiterer wichtiger Unterschied besteht darin, dass nur durch entsprechende Prozesssteuerung sehr hohe Mutationsraten, die dem 10.000- bis Million-fachen der natürlichen Rate entsprechen, eine genügend große Anzahl von Mutationen erzeugt werden kann, um unter diesen die seltenen (eine einzige unter einer Milliarde) positiven Mutanten zu finden. Außerdem muss ein schnelles und effektives Screening erfolgen, damit die seltenen positiven Mutanten erkannt und ausgelesen werden können. Nicht zuletzt funktioniert das Ganze nur mit sorgfältig ausgewählten Reaktionsbedingungen, dem intelligent ausgewählten Einsatz von gentechnischen Werkzeugen (Werkzeuge, die selbst intelligent gestaltet sind) und einer optimalen Auswahl von Varianten für das gewünschte Ziel. Nicht umsonst wurde nun für diese bahnbrechenden Forschungen ein Nobelpreis vergeben, und zwar sicher nicht dafür, dass nur ein an sich geist- und zielloser Prozess – natürliche Evolution – nachgeahmt wurde. Damit kommen wir zu einem weiteren Punkt.
Begriffliches Verwirrspiel. Der Begriff „gerichtete Evolution“ ist ein Widerspruch in sich (contradictio in adjecto), so wie zum Beispiel „hölzernes Eisen“ und täuscht etwas vor, das es gar nicht gibt. Denn mit Evolution (im stammesgeschichtlichen Sinne) ist ein Prozess gemeint, der ohne Zielvorgabe und Steuerung verläuft, eben ungerichtet. Das liegt daran, dass Mutationen ungerichtet sind, aber auch die Selektionsbedingungen keinem Ziel folgen und nicht geplant sind. Evolution kann aber nicht zugleich gerichtet und ungerichtet sein. Letztlich sind Proteine, die durch „gerichtete Evolution“ entstanden sind, durch intelligentes Design entwickelt worden, wobei der Mutationsprozess und zielorientierte Auslese intelligent eingesetzt werden. Nicht umsonst wird auch von „bioengineering“ gesprochen.
Mit dem Begriff „gerichtete Evolution“ soll ein Unterschied zum sogenannten „rationalem Design“ zum Ausdruck gebracht werden. Damit bezeichnet man die Strategie, gezielt Moleküle zu synthetisieren, auf der Basis der Kenntnisse über dieses Molekül. Auch „rationales Design“ impliziert einen Widerspruch in sich, da er suggeriert, es gebe auch „nichtrationales Design“; das aber wäre ebenfalls ein „hölzernes Eisen“ und widerspricht der üblichen Bedeutung von „Design“.
Grenzen gerichteter Evolution. Änderungen erfolgen nur, wenn einzelne Mutationen (seien sie künstlich erzeugt oder in der Natur auftretend) bereits einen Vorteil ermöglichen und daher ausgelesen werden können. Die Entstehung neuer Proteindomänen oder neuer Proteinfolds erfordert jedoch gemäß experimenteller Daten zahlreiche Änderungen gleichzeitig. Der Biochemiker Michael Behe hat in seinem Buch „The Edge of Evolution“ (Behe 2007) aufgrund von Forschungen zu E. coli-, Malaria- und HIV-Mutationen gezeigt, dass Änderungen auch in evolutionären Zeiträumen nicht zu erwarten sind, wenn nur drei (oder mehr) passende Zufallsmutationen gleichzeitig auftreten müssten.3 Wichtig ist also: Bei gerichteter Evolution geht es um Optimierung, nicht um Innovationen.
Tatsächlich können die Wissenschaftler mit gerichteter Evolution in Bezug auf Optimierung mehr erreichen als die Natur. Beispielsweise produzierte Arnold eine Variante eines proteinabbauenden Enzyms mit 10 Mutations- und Selektionszyklen, das in einem organischen Lösungsmittel mehr als 200-mal stabiler war als die natürliche Variante. Wissenschaftler können mehr erreichen als zukunftsblinde Evolution, weil sie sich für ein Ziel entscheiden: In der ersten Runde können sie ein mutiertes Gen mit z. B. zwei gleichzeitig auftretenden Mutationen erhalten. Wenn dieses neue Gen ausgewählt und mutiert wird, können zwei weitere Mutationen und damit insgesamt vier Mutationen im ursprünglichen Gen erhalten werden. Doch all das betrifft – um es nochmals zu betonen – die Optimierung einer bestehenden Struktur bzw. eines funktionalen Proteins – nicht die Schaffung qualitativ neuer Strukturen oder Proteine.4
Dank. Ich danke Prof. Dr. Matti Leisola, der selbst auf dem Gebiet der Biotechnologie gearbeitet hat, und Dr. Harald Binder für wertvolle Hinweise. Matti Leisola hat ebenfalls eine Stellungnahme zur Vergabe des Nobelpreises verfasst: https://evolutionnews.org/2018/10/how-the-2018-nobel-laureates-in-chemistry-harnessed-intelligent-design/
Weitere Stellungnahme des Discovery-Instituts: https://evolutionnews.org/2018/10/its-not-evolution-a-nobel-prize-award-for-engineering-enzymes/
Anmerkungen
[1] https://www.wissenschaft.de/gesundheit-medizin/chemie-nobelpreis-fuer-gelenkte-evolution
[2] Z. B.: „Sie bauen die Natur im Schnelldurchlauf nach“ (https://www.zeit.de/wissen/2018-10/nobelpreis-in-chemie-fuer-proteinforscher-frances-arnold-george-smith-und-sir-gregory-winter); „die Prinzipien der biologischen Evolution für die Entwicklung neuer Enzyme … nutzbar gemacht“ (http://www.scinexx.de/newsletter-wissen-aktuell-23225-2018-10-04.html); „Demnach baut ihr Werk auf niemand Geringerem auf als auf Charles Darwin, dem Begründer der Evolutionstheorie“ (https://www.sueddeutsche.de/wissen/nobelpreis-fuer-chemie-so-kam-die-evolution-ins-reagenzglas-1.4154859)
[3] Vgl. Romero PH & Arnold FH (2009) Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 10, 866-875. „Man muss sehr lange warten, bis vier oder fünf spezifische Mutationen auftreten, die eine gewünschte Funktion erfüllen. In einer Bakterienpopulation wie E. coli kann die Wartezeit auf vier Mutationen 1015 Jahre betragen, wenn man die global anzunehmenden Populationsgrößen zugrunde legt. (Das Universum ist nach üblicher Auffassung nur 1014 Jahre alt.)“ (in Übersetzung); siehe auch: Reeves MA, Gauger AK & Axe DD (2014) Enzyme families — Shared evolutionary history or shared design? A study of the GABA-aminotransferase family. BIO-Complexity 2014 (4):1−16.
[4] Mit den Möglichkeiten der Wissenschaftler im Vergleich zu natürlichen Vorgängen befassen sich B. Kozulik & M. Leisola im Artikel „Have Scientists Already Been Able to Surpass the Capabilities of Evolution?“ (http://vixra.org/abs/1504.0130). Sie schreiben im Vorwort: „Die im Titel dieser Arbeit gestellte Frage wird für manche Leser paradox, rätselhaft oder einfach dumm klingen. Sie werden denken: Wenn Wissenschaftler wie alle anderen lebenden Organismen das Produkt der Evolution sind, wie konnten sie dann ihre „Fähigkeiten“ überbieten, wenn wir doch nicht einmal in der Lage sind, einen einzigen wirklich neuen lebenden Organismus zu produzieren? Wie kann es wahr sein, dass die Wissenschaftler, nachdem sie nur eine kurze Strecke auf dem Weg zur Schaffung eines neuen lebenden Organismus zurückgelegt haben, bereits mehr als die Evolution leisten konnten? Und wenn eine Gruppe von Wissenschaftlern dies tatsächlich erreicht hat, wie kommt es dann, dass sie für ihren Publikationen keine Anerkennung für eine so große Leistung erhalten haben? Das Ziel dieses Artikels ist es, zu zeigen, dass die Antwort auf die Titelfrage „ja“ ist.“
Autor dieserNews: Reinhard Junker
© 2018, http://www.genesisnet.info/schoepfung_evolution/n261.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
12.07.18 Vom Salzwasser- zum Süßwasserfisch?
In Nordamerika heimische Stahlkopfforellen leben normalerweise sowohl in Süßwasser als auch zeitweise in Salzwasser und können ihre Osmoregulation entsprechend umschalten. Eine Population hat sich binnen 100 Jahren auf das Leben ausschließlich im Süßwasser spezialisiert. Die damit einhergehenden Änderungen des Erbguts erfolgten in kurzer Zeit, was nur auf der Basis einer ursprünglich großen Variationsbreite möglich war.
Stahlkopfforellen (Oncorhynchus mykiss) liefern ein interessantes Beispiel zur programmierten Variabilität von Organismen. Diese in Nordamerika von Alaska bis Kalifornien beheimateten Fische sind in der Lage, sowohl in Salzwasser als auch in Süßwasser zu leben. Sie schlüpfen in Süßwasser, verbleiben 1-2 Jahre dort, wandern dann über Flüsse in den pazifischen Ozean und verbringen dort 1-3 Jahre, um zur Laichzeit ins Süßwasser zurückzukehren.
Die Erfordernisse bezüglich der Osmoregulation (Gleichgewicht zwischen Salzen und Ionen innerhalb der Zellmembranen) sind im Süß- und Salzwasser gegensätzlich. Im Süßwasser müssen die Fische in der Lage sein, aktiv Ionen aus ihrer Umwelt aufzunehmen, um so den Salzverlust durch passive Diffusion auszugleichen, während im Salzwasser Ionen abgegeben werden müssen, um die Salzaufnahme durch ihren Körper auszugleichen. Fische, die in beiden Medien leben, benötigen also beide Mechanismen zur Aufrechterhaltung der erforderlichen Salzkonzentration in den Zellen und können zwischen ihnen umschalten. Darüber hinaus unterscheidet sich der Lebensraum im Süßwasser von der Salzwasserumgebung auch in der Wassertemperatur, in der Art der Strömung und in der Zusammensetzung der sonstigen Fauna.
Vor 120 Jahren wurden einige Stahlkopfforellen im Lake Michigan ausgesetzt. Nach anfänglichen Schwierigkeiten konnten die Fische eine stabile Population aufbauen. Für diese Fische war der große See gleichsam ein Ozean-Ersatz; sie wanderten nicht ins Meer, sondern verblieben dauerhaft im Süßwasser. Nun haben Forscher das Genom (komplettes Erbgut) von 264 Exemplaren der Stahlkopfforelle genetisch untersucht und eine Reihe von Unterschieden zwischen den Süßwasserformen und den in den Pazifik wandernden Formen entdeckt (Willoughby et al. 2018). Dabei zeigte sich, dass zwei veränderte Chromosomenregionen in einer direkten Verbindung mit der Osmoregulation stehen: Die Carboanhydrase (katalysiert die Hydratisierung von Kohlenstoffdioxid zu Kohlensäure und umgekehrt) und ein Transportprotein (SLC 26) sind in einer Weise verändert, dass die Ionenaufnahme aus dem Wasser erleichtert ist. Die oben erwähnte Fähigkeit, die Osmoregulation bei Süß- und Salzwasser umschalten zu können, ist energetisch kostspielig, so dass der Verlust dieser Fähigkeit bei dauerhafter Existenz im Süßwasser zudem von Vorteil ist (Willoughby et al. 2018, 8).
Die dritte veränderte Genregion betrifft das Enzym Ceramid-Kinase, das im Zusammenhang mit dem Stoffwechsel bei der Wundheilung der Tiere steht. Die im Süßwasser lebenden Tiere sind vermehrt Verletzungen durch Neunaugen ausgesetzt und Wunden heilen im Süßwasser schlechter; durch die Veränderung wird vermutlich der Heilungsprozess verbessert (Willoughby et al. 2018, 8).
Wie konnten sich die Gene aber so schnell verändern? Die Forscher fanden heraus, dass die Veränderungen weder auf Hybridisierungen mit anderen Stahlkopfforellen-Populationen noch auf Mutationen zurückgeführt werden konnten. Vielmehr sprechen die Befunde dafür, dass die anfangs überlebenden Individuen bereits die passenden Ausprägungen der relevanten Gene besaßen und daher am ehesten überleben konnten. Die spezialisierten Forellen des Lake Michigan „leben“ also von einer ursprünglichen genetischen Vielseitigkeit ihrer Vorläufer.
Bemerkenswert ist eine weitere Beobachtung: Die Wissenschaftler stellten fest, dass die genetische Vielfalt der Stahlkopfforellen im neuen Lebensraum in allen 29 Chromosomen deutlich niedriger liegt als bei den Populationen, die in den angestammten Gewässern leben, was auf einen Gründer-Effekt zurückgeführt wird. Spezialisierung ist wie in vielen anderen Fällen mit genetischer Verarmung verbunden.
Diskussion. Das Beispiel der Osmoregulation bei den Stahlkopfforellen reiht sich in eine lange Liste von Fällen ein, in denen von einer ursprünglich genetisch vielseitigen (polyvalenten) Ausgangssituation ausgegangen werden muss – soweit diese rekonstruiert werden kann – und in denen eine schnelle Veränderung beobachtet wurde (in Verlauf von Jahren bis Jahrzehnten). Die beobachteten Veränderungen und Spezialisierungen gehen mit Einschränkung der genetischen Vielfalt einher. Die ursprüngliche Vielseitigkeit kann enorm sein; immerhin ist es keine Kleinigkeit, sowohl im Süßwasser als auch im Salzwasser leben zu können.
Auf der Basis dieser Befunde kann man spekulieren, dass eine ursprünglich große Toleranz bezüglich des Salzgehalts des Wassers bei heute weniger toleranten Arten sozusagen eine eingefrorene Spezialisierung ist (verbunden mit genetischer Verarmung). Eine große Toleranzbreite könnte auch bei den umwälzenden Prozessen während der Sintflut eine wichtige Voraussetzung für das Überleben gewesen sein.
Literatur
Willoughby JR, Harder AM, Tennessen JA, Scribner KT & Christie MR (2018) Rapid genetic adaptation to a novel environment despite a genome‐wide reduction in genetic diversity. Mol. Ecol., doi:10.1111/mec.14726
Autor dieserNews: Reinhard Junker
© 2018, http://www.genesisnet.info/schoepfung_evolution/n260.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
23.12.16 Entstehung evolutionärer Neuheiten – ungelöst!
Zentrale Fragen zur Aufklärung der Entstehung evolutionärer Neuheiten und ihrer Ursachen sind unbeantwortet. Die zunehmende Kenntnis über die Details lebender Organismen und ihrer Konstruktionen lässt die Unzulänglichkeit der bisherigen kausalen Evolutionstheorien umso deutlicher hervortreten. Die Kontroverse darüber wird innerhalb der Evolutionsbiologie offen ausgetragen.
In populären Darstellungen über Evolution, in Schul- und Lehrbüchern oder auch in interdisziplinär-theologischen Abhandlungen wird schon seit Jahrzehnten behauptet, dass ein evolutionärer Ursprung der Lebewesen eine Tatsache sei. Darüber hinaus könne auch als geklärt gelten, dass und wie Evolution nach rein natürlichen Mechanismen – d. h. ohne zielgerichteten, schöpferischen Input – abgelaufen sei. In einem kirchlichen Text wird folgende Einschätzung gegeben: „Die Frage ist, ob das Wechselspiel von genetischen Veränderungen und Selektion eine hinreichende naturwissenschaftliche Erklärung für die Evolution der irdischen Lebenswelt liefert! Die gegenwärtige Biologie beantwortet diese Frage mit Ja“ (Hemminger 2007, 22).
Eine wachsende Zahl von Evolutionsbiologen würde dieser Einschätzung bezüglich den „Wie“ allerdings nicht zustimmen. Im Jahr 2003 listeten der Wiener Entwicklungsbiologe Gerd B. Müller und der Zellbiologe und Anatom Stuart A. Newman eine große Anzahl ungelöster Fragen der kausalen Evolutionsforschung auf (vgl. Mikroevolution, Makroevolution und „ID“, https://www.genesisnet.info/index.php?News=93); die ersten sechs davon lauten:
- Burgess shale-Effekt: Weshalb entstanden die Baupläne der Vielzeller explosionsartig?
- Homoplasie: Weshalb entstehen ähnliche Gestalten unabhängig und wiederholt?
- Konvergenz: Weshalb produzieren entfernt verwandte Linien ähnliche Designs?
- Homologie: Weshalb organisieren sich Bauelemente als fixierte Baupläne und Organformen?
- Neuheit: Wie werden neue Elemente in bestehende Baupläne eingeführt?
- Modularität: Weshalb werden Design-Einheiten wiederholt verwendet?
Es ist leicht zu sehen, dass es sich nicht um Randfragen oder spezielle Details handelt, sondern um zentrale Fragen der evolutionsbiologischen Forschung: Wie können Neuheiten und die Organisation der Lebewesen entstehen? In der Folgezeit wurde in Fachartikeln von verschiedenen Autoren immer wieder zum Ausdruck gebracht, dass diese Fragen nach wie vor auf Antworten warten. Nachfolgend einige Beispiele (in Übersetzung des Autors; die z. T. längeren Originalzitate finden sich im Anhang:
- „Die Prozesse, die evolutionären Innovationen zugrunde liegen, sind bemerkenswert wenig verstanden. … [W]ir haben relativ wenig Fortschritt erzielt im Verständnis, wie neue Eigenschaften erstmals entstehen“ (Moczek 2008, 432).
- „Eines der bedeutendsten ungelösten Probleme der Biologie ist das Verständnis, wie neue, komplexe Phänotypen [= Gestalten] entstehen, sowohl in der individuellen Entwicklung als auch in der Stammesgeschichte“ (Ledon-Rettig et al. 2008, 316).
- „Die Skelettarchitektur von Wirbeltieren ist sehr unterschiedlich, doch die Basis für Veränderungen im groben Skelettbau bleibt fast völlig unbekannt“ (Rudel & Sommer 2003, 21).
- „[D]ie Kernfrage – der genetische Ursprung neuer und komplexer Merkmale – ist wahrscheinlich eine der hartnäckigsten und fundamentalen unbeantworteten Fragen in der Evolutionsforschung heute“ (Monteiro & Podlaha 2009, 215).
- „[D]ie genetischen Änderungen, die erklären, wie komplexe Formen entstehen, sind immer noch unklar“ (Martin et al. 2012, 12632).
- „Der Ursprung und die Diversifizierung neuer Eigenschaften ist eine der spannendsten ungelösten Probleme in der evolutionären Entwicklungsbiologie“ (Saenko et al. 2011, 1).
- „Wie Körperbaupläne in der Natur evolvieren, bleibt weitgehend unbekannt“ (Cleves et al. 2014, 13912).
- „Wir sind uns noch nicht sicher, welches die Erklärung für Neuheiten ist. … Ich vermute, dass der Ursprung von Neuheiten auch natürliche Selektion benötigt, ebenso wie zusätzliche Mechanismen, aber worin diese bestehen, muss erst noch durch weitere empirische Forschung herausgefunden werden“ (Wagner 2014, 125).
Im Jahr 2010 veröffentlichten Jerry Fodor und Massimo Piattelli-Palmarini das viel beachtete und auch viel gescholtene Buch „What Darwin got wrong“. Darin behaupten (und begründen) sie, dass (und warum) die Selektionstheorie darin scheitere, die Entstehung neuer Formen zu erklären, und dass es derzeit auch keine alternative Erklärung gebe ([vgl. Lag Darwin falsch?, https://www.genesisnet.info/index.php?News=153). Als bekennende Atheisten sind sie aber überzeugt, dass es eine rein naturalistische Erklärung geben muss, die derzeit jedoch nicht bekannt sei. (Für seine Kritik wurde Jerry Fodor von David Sloan Wilson als „säkularer Kreationist“ bezeichnet, siehe http://www.suzanmazur.com/?p=20)
Und der in den USA bekannte und renommierte atheistische Philosoph Thomas Nagel veröffentlichte im Jahr 2012 das Buch „Mind and Cosmos“, in Deutsch 2013 veröffentlicht mit dem Untertitel „Warum die materialistische neodarwinistische Konzeption der Natur so gut wie sicher falsch ist“. Darin thematisiert Nagel u. a. die „immensen Schwierigkeiten, die Entstehung des Lebens und der Arten allein durch nichtgerichtete Prozesse verständlich zu machen“ (Widenmeyer 2013).
Die Kontroverse wird offen ausgetragen. Die Zahl der Kritiker der Standard-Evolutionstheorie (Neodarwinismus, Synthetische Evolutionstheorie, Erweiterte Synthese u. a.) war schließlich so stark angeschwollen, dass die bedeutende Wissenschaftszeitschrift Nature im Oktober 2014 unter der Überschrift „Does evolutionary theory need a rethink?“ ein Pro & Contra von zwei Wissenschaftler-Gruppen veröffentlichte (deutsche Übersetzung: http://www.spektrum.de/news/brauchen-wir-eine-neue-evolutionstheorie/1320620). Die eine Gruppe plädierte für ein „dringendes Ja“, also für ein Umdenken (Laland et al. 2014), während für die andere Gruppe alles in bester Ordnung ist (Wray et al. 2014) (vgl. „Brauchen wir eine neue Evolutionstheorie?“, https://www.genesisnet.info/index.php?News=221).
Der letzte Höhepunkt dieser Entwicklung war eine wissenschaftliche Tagung der britischen Royal Society Anfang November 2016, auf der die Kontroverse zwischen den beiden Lagern drei Tage lang ausgetragen wurde. Siegfried Scherer berichtet: „Die Kritiker meinen, dass der Neodarwinismus ‚dringend überdacht‘ werden müsse, unter anderem um Höherentwicklung im Evolutionsprozess zu erklären. Die Neodarwinisten halten dagegen, dass die Kritiker eigentlich nichts grundsätzlich Neues vorzutragen hätten, mit der herrschenden Lehre sei ‚alles gut‘.“ Es stand nicht weniger als die Frage im Raum, ob alle bisher vorgeschlagenen Erklärungsversuche unzureichend sein könnten (Bericht unter https://www.pro-medienmagazin.de/kultur/veranstaltungen/detailansicht/aktuell/hatte-darwin-doch-nicht-recht-98445/). Die Streitpunkte waren dieselben wie beim o. g. Pro & Contra in Nature – in Kurzform:
- Der genzentrierte Ansatz (Genänderungen als Initialzündungen) müsse ergänzt werden durch die Berücksichtigung zahlreicher Wechselwirkungen mit äußeren und inneren Einflüssen während der ontogenetischen Entwicklung.
- Die Umweltbedingungen haben nicht nur eine passive Rolle als Selektionsfaktoren, vielmehr werde die Umwelt durch die Lebewesen aktiv mitgestaltet (Nischenkonstruktion); dadurch beeinflussen die Lebewesen selbst auch ihre eigene Evolution.
- Durch die Plastizität der Lebewesen (Änderungen infolge von Umweltreizen – ohne Genänderungen) sei eine schnelle Anpassung und sogar die Offenlegung bisher verborgener Merkmale möglich, die nachfolgend durch Genvariationen (Mutationen) dauerhaft sichtbar fixiert werden können.
- Extragenetische Veränderungen (Epigenetik) in der Gen-Regulation könnten wie die Gene selber ebenfalls vererbt werden und Einfluss auf Evolution nehmen.
Im Beitrag „Brauchen wir eine neue Evolutionstheorie?“ (https://www.genesisnet.info/index.php?News=221) wird erläutert, warum diese Vorschläge für die Erklärung evolutionärer Neuheiten nicht zielführend sind.
Fazit. Von einer Klärung der Evolutionsmechanismen, die zu neuartigen Bauplänen von Organismen führen, kann nicht die Rede sein – im Gegenteil: Je mehr die Details lebender Konstruktionen aufgeklärt werden, desto deutlicher zeigt sich die Unzulänglichkeit der bisherigen kausalen Evolutionstheorien. In Bezug auf die Frage nach den Mechanismen der Makroevolution – der Entstehung von Neuheiten – hat der Wissensfortschritt die Erklärungsdefizite nicht kleiner gemacht, sondern deutlicher hervortreten lassen. Solange die Diskussion sich freilich nur im Rahmen naturalistischer Antwortmöglichkeiten bewegt und – ohne klare Sachargumente und ohne zwingende methodologische Begründung – die Option „Schöpfung“ aus dem Rennen um die zutreffende Antwort herausgehalten wird, kann es – allein aus wissenschaftstheoretischer Sicht – sein, dass nur um die beste Hypothese im Rahmen eines falschen Ansatzes gestritten wird.
Literatur
Cleves PA, Ellis NA, Jimenez MT, Nunez SM, Schluter D, Kingsley DM & Miller CT (2014) Evolved tooth gain in sticklebacks is associated with a cis-regulatory allele of Bmp6. Proc. Natl. Acad. Sci. 111, 13912-13917.
Fodor J & Piattelli-Palmarini (2010) What Darwin got wrong. News York
Hemminger H (2007) Mit der Bibel gegen die Evolution. EZW-Texte 195.
Ledon-Rettig CC, Pfennig DW & Nascone-Yoder H (2008) Ancestral variation and the potential for genetic accommodation in larval amphibians: implications for the evolution of novel feeding strategies. Evol. Dev. 10, 316-325.
Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G, Jiggins CD, Kronforst MR, Long AD, McMillan WO & Reed RD (2012) Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc. Natl. Acad. Sci. 109, 12632-12637.
Moczek AP (2008) On the origins of novelty in development and evolution. BioEssays 30, 432-447.
Monteiro A & Podlaha O (2009) Wings, horns, and butterfly eyespots: How do complex traits evolve? PLoS Biology 7:2, 0209-0216; doi: 10.1371/journal.pbio.1000037.
Müller GB & Newman SA (2003) Origination of organismal form: The forgotten cause in evolutionary theory. In: Müller GB & Newman SA (eds) Origination of organismal form. Beyond the gene in developmental and evolutionary biology. Vienna Series in Theoretical Biology. Cambridge, MA, pp 3-12.
Laland K et al. (2014) Does evolutionary theory need a rethink? Yes, urgently. Nature 514, 161-164.
Moczek AP (2008) On the origins of novelty in development and evolution. BioEssays 30, 432-447.
Rudel D & Sommer RJ (2003) The evolution of developmental mechanisms. Dev. Biol. 264, 15-37.
Saenko SV, Maralva MSP & Beldade P (2011) Involvement of the conserved Hox gene Antennapedia in the development and evolution of a novel trait. EvoDevo 2011, 2:9; doi: 10.1186/2041-9139-2-9.
Wagner GP (2014) Homology, genes, and evolutionary innovation. Princeton University Press.
Widenmeyer M (2013) Rezension von: Geist und Kosmos. Warum die materialistische neodarwinistische Konzeption der Natur so gut wie sicher falsch ist. Studium Integrale Journal 20, 124-126. http://www.wort-und-wissen.de/sij/sij202/sij202-r1.html
Wray G et al. (2014) Does evolutionary theory need a rethink? No, all is well. Nature 514, 161-164.
Anhang: Originalzitate
„Given its importance and pervasiveness, the processes underlying evolutionary innovation are, however, remarkably poorly understood, which leaves us at a surprising conundrum: while biologists have made great progress over the past century and a half in understanding how existing traits diversify, we have made relatively little progress in understanding how novel traits come into being in the first place“ (Moczek 2008, 432).
„One of biology’s most significant unresolved issues is to understand how novel, complex phenotypes originate, both developmentally and evolutionarily“ (Ledon-Rettig et al. 2008, 316),
„The skeletal architecture of vertebrates is widely divergent, yet the basis for change in gross skeletal morphology remains almost entirely unknown“ (Rudel & Sommer 2003, 21).
„This work is difficult and time consuming, but the question at its core—the genetic origin of new and complex traits—is probably still one of the most pertinent and fundamental unanswered questions in evolution today“ (Monteiro & Podlaha 2009, 215).
„Although animals display a rich variety of shapes and patterns, the genetic changes that explain how complex forms arise are still unclear“ (Martin et al. 2012, 12632).
„The origin and diversification of novel traits is one of the most exciting unresolved issues in evolutionary developmental biology“ (Saenko et al. 2011, 1).
„How body pattern evolves in nature remains largely unknown“ (Cleves et al. 2014, 13912).
„The explanation for adaptation is natural selection. We are not yet sure what the explanation for novelties is. … I suspect that the origin of novelties also requires natural selection as well as additional mechanisms, but what they are will have to be determined by more empirical research“ (Wagner 2014, 125).
Autor dieserNews: Reinhard Junker
© 2016, http://www.genesisnet.info/schoepfung_evolution/n243.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
21.06.16 Birkenspanner, Melanismus und springende Gene
Durch Analyse des Erbguts des berühmten Birkenspanners (Biston betularia) und den Vergleich mit dem Genom des Seidenspinners (Bombyx mori) kann das Lehrbuchbeispiel des Melanismus besser verstanden werden. Die dunklen und hellen Erscheinungsformen des Spanners werden durch sogenannte „springende Gene“ verursacht. Die Untersuchungen deuten darauf hin, dass sich die genetische Veränderung erst kurz vor dem auffällig häufigen Vorkommen der dunklen Varianten ereignet hat.
Der Birkenspanner (Biston betularia) ist ein häufig verwendetes Lehrbuchbeispiel dafür, wie aufgrund von Umweltbedingungen unterschiedliche Erscheinungsformen von Lebewesen (Morphen oder Phänotypen) selektiert werden können. Die hellen und dunklen Morphen von B. betularia können z. B. von Fressfeinden auf hellem bzw. dunklem Untergrund unterschiedlich gut wahrgenommen und erbeutet werden. Obwohl der Einfluss und die Reichweite von Selektionsfaktoren bis in die Gegenwart kontrovers diskutiert werden, ist hinreichend belegt, dass Vögel als Fressfeinde die Häufigkeitsverteilung der Birkenspanner in Abhängigkeit des entsprechenden Untergrundes beeinflussen.

Abb. 1: Paarung der beiden Birkenspanner-Morphen (hell und dunkel) von Biston betularia. (Wikimedia: Siga, CC BY-SA 4.0)
Die genetischen Grundlagen für das Auftreten verschiedener Phänotypen von B. betularia sind bisher allerdings erst in Ansätzen bekannt. Van´t Hof et al. (2011) konnten den Ort der genetischen Ursache für das Auftreten der dunklen Morphe durch einen Vergleich mit dem Seidenspinner (Bombyx mori) anhand von genetischen Markern auf dem Chromosom 17 auf einen Bereich von 200 Kilobasen (kb) eingrenzen. Die Autoren interpretierten die empirischen Befunde als Beleg dafür, dass ein Allel (Genvariante), das vor nicht langer Zeit („recent“) erstmals aufgetreten ist, für die dunklen Birkenspanner verantwortlich ist. Sie konnten damals zwar die Korrelation des Bereichs im Genom mit der farblichen Veränderung belegen, aber es war kein genetischer Zusammenhang mit der Melaninproduktion, also der Erzeugung des dunklen Pigments, erkennbar.
In einer neuen Untersuchung (Van´t Hof et al. 2016) konnte nun nachgewiesen werden, dass die dunklen B. betularia-Formen durch ein springendes Gen (Transposon) verursacht werden. In diesem Fall haben die Autoren ein Transposon der Klasse II identifiziert, also einen mobilen DNA-Abschnitt, der seine Position im Genom selbständig verändern kann. In der Arbeit wird gezeigt, dass dieses Transposon sich in dem zuvor beschriebenen Bereich des Chromosoms 17 in das erste Intron eines Gens mit der Bezeichnung cortex integriert. Das Transposon umfasst einen DNA-Strang mit 22 kb, der sich in einen Abschnitt des cortex-Gens einbaut, der vor der Übersetzung (Translation) in das entsprechende Protein herausgeschnitten wird. Vom Protein Cortex ist aber nicht bekannt, dass es in irgendeinem Zusammenhang mit der Biosynthese des dunklen Pigments Melanin steht. Das cortex-Gen wird in bestimmten Larvenstadien in den embryonalen Flügelanlagen stark ausgeprägt (exprimiert). Das entsprechende Protein Cortex reguliert den Zellzyklus während der Entwicklung der Flügel in der Larve. Die genauen Details, wie diese Regulation die Pigmentierung beeinflusst, sind bisher jedoch nur wenig verstanden und werden weiter erforscht. In einem gleichzeitig erschienenen Artikel bestätigen Nadeau et al. (2016) jedoch, dass cortex in Schmetterlingen die Musterung der Flügel kontrolliert.
Aufgrund statistischer Untersuchungen von cortex in Birkenspannern kommen Van´t Hof et al. (2016) zu dem Schluss, dass der Zeitpunkt, an dem das springende Gen sich an der neuen Position integriert hat, um das Jahr 1819 liegen soll. Sollte sich das bestätigen, dann hätte sich diese genetische Veränderung sehr rasch in der Population der Birkenspanner in der Gegend von Birmingham bemerkbar gemacht, denn Mitte des 19. Jahrhunderts wurden die dunklen Morphen dort beobachtet und beschrieben.
Wenn sich die hier vorgestellten Untersuchungsergebnisse und deren Interpretation durch weitere Forschung bestätigen sollten, dann würde dieser Fall von Industriemelanismus zeigen, dass die verschiedenen Erscheinungsformen des Birkenspanners quasi vorprogrammiert sind und sich in einer Population rasch etablieren können.
Literatur
Van´t Hof AE, Edmonds N, Dalikova M, Marec F & Saccheri IJ (2011) Peppered moths has a singular and recent mutational origin. Science 332, 958-960.
Van´t Hof AE, Campagne P et al. (2016) The industrial melanism mutation in British peppered moths is a transposable element. Nature 534, 102-105.
Nadeau NJ, Pardo-Diaz C et al. (2016) The gene cortex controls mimicry and crypsis in butterfly and moths. Nature 534, 106-110.
Autor dieserNews: Harald Binder
© 2016, http://www.genesisnet.info/schoepfung_evolution/n238.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
28.01.14 Geheimnisvolle DNA – überlagernde Codes im Genom
Die Hinweise mehren sich, dass in der DNA, in der das Genom* niedergelegt ist, dem „universellen“ genetischen Code für die Biosynthese von Proteinen verschiedene weitere Codes überlagert sind. Jüngst wurden experimentelle Befunde veröffentlicht, die zeigen, dass die Bindung von bestimmten Proteinen (sog. Transkriptionsfaktoren) an die DNA einen Einfluss darauf hat, welche Nukleotide in den Codons** verwendet werden.1 Ein und dieselbe DNA-Sequenz wird in diesen Fällen also auf mindestens zwei ganz verschiedene Weisen genutzt.
*Genom (=gesamtes Erbgut eines Individuums)
**Codon (= Abfolge von drei Nukleobasen (= Triplett), die eine Aminosäure codiert)
Nachdem Watson & Crick (1953) die Struktur der Doppelhelix für die DNA veröffentlicht hatten, wurde in den folgenden Jahren viel Forschung in die Entschlüsselung des genetischen Codes investiert. Das Schlüsselexperiment gelang Heinrich Matthaei im Labor von Marshall Nirenberg am 27. Mai 1961. Die vollständige Zuordnung der 64 Nukleotidtripletts zu den 20 Aminosäuren, die für Proteine als Baustein verwendet werden, gelang bis 1966 vor allem auch durch die Beiträge aus der Arbeitsgruppe von Khorana. Die vor allem in den letzten Jahren mit der Erforschung des menschlichen Genoms gesammelte Erfahrung, dass nur ein vergleichsweise kleiner Teil des Genoms für Proteine codiert, verstärkte die Fragen nach weiteren Funktionen des Genoms.
Inzwischen ist klar, dass manche Proteine spezifisch an DNA binden und dadurch die Verfügbarkeit der dort codierten Information regulieren. Auch die Wechselwirkung von DNA-Sequenzen mit verschiedenen RNA-Molekülen beeinflusst die Aktivität von Genen. Weateritt & Babu (2013) sind der Meinung, dass die Bindungen von Proteinen und RNA an bestimmte genomische DNA-Sequenzen als unabhängige zusätzliche Codes betrachtet werden können. Beispielsweise untersuchten Sterachis et al. (2013) die Bindung eines bestimmten Typs von Proteinen, sogenannten Transkriptionsfaktoren, an die DNA des menschlichen Genoms in 81 verschiedenen Zelltypen. Sie konnten zeigen, dass etwa 15 % der Codons eines Gens (also den Basentripletts, die sonst für eine Aminosäure codieren) in rund 87 % der menschlichen Gene auch in die Bindung mit Transkriptionsfaktoren einbezogen werden. Die Autoren bezeichnen diese Genbereiche als „Duone“ und bringen damit zum Ausdruck, dass sie Information auf zwei Weisen codieren: einmal „klassisch“ für Proteine und zusätzlich durch die Bindung der Transkriptionsfaktoren für Regulation der Gene.
Die Wechselwirkung eines Proteins mit einer DNA-Sequenz, die gleichzeitig ein Protein codiert, stellt besondere Anforderungen an die genutzten Codons und die dadurch festgelegten Aminosäuren. Sterachis et al. untersuchten die Auswirkung des Austausches einzelner Nukleotide in Duonen auf die Fähigkeit, Transkriptionsfaktoren zu binden. Zunächst stellten sie dazu die bekannten genetischen Varianten zusammen, die mit Krankheiten oder mit einer veränderten Funktion der Duonen in Zusammenhang gebracht werden. Mehr als 17 % der Duonen, in denen einzelne Nukleotide ausgetauscht waren, zeigten eine veränderte Bindung der Transkriptionsfaktoren. Der Austausch von synonymen Codons, die also für dieselbe Aminosäure in den Proteinen codieren, führt zwar zum selben funktionsfähigen Protein, jedoch zu einer Veränderung der Bindung der Transkriptionsfaktoren. Dieser Befund deutet darauf hin, dass einige Änderungen in Duonen hauptsächlich ihre Transkriptionsfaktor-Bindung beeinflussen. Änderungen in der DNA-Sequenz können somit offensichtlich auch dann Krankheiten verursachen, wenn sie gar nicht zu Modifikationen in Proteinen führen. Der Austausch einzelner Nukleotide (single nucleotide polymorphism; snp) im Genom kann also auf vielfältige Weise einen Organismus beeinflussen; das gesamte Spektrum von regulatorischen Codes muss somit in Betracht gezogen werden.
Die Erkenntnis, dass im Genom verschiedene Codes enthalten sind und genutzt werden, eröffnet Einblicke in eine „Vieldimensionalität“ des Genoms. Diese erst ansatzweise verstandene Komplexität des Erbguts kann zum Staunen führen und eröffnet völlig neue Fragestellungen. Bisherige Vorstellungen zur Entstehung der im Genom abgespeicherten Information müssen neu überdacht werden, da die DNA-Sequenzen mit einer bisher ungeahnten Vielfalt an Codes abgelesen werden (s. z. B. auch Fellner et al. 2014).
Literatur
Fellner L, Bechtel N, Witting MA, Sion S, Schmitt-Kopplin P, Keim D, Scherer S & Neuhaus K (2014) Phenotype of htgA (mbiA), a recently evolved orphan gene Escherichia coli and Shigella, completely overlapping in antisense to yaaW. FEMS Microbiol. Lett. 350, 57-64.
Stergachis AB, Haugen E, Shafer A, Fu W, Vernot B, Reynold A, Raubitschek A, Ziegler S, LeProusr EM, Akey JM & Stamatoyannopoulos JA (2013) Exonic transcription factor binding directs codon choice and affects protein evolution. Science 342, 1367-1372.
Weatheritt RJ & Babu MM (2013) The hidden codes that shape protein evolution. Science 342, 1325-1326.
Watson JD & Crick FHC (1953) Molecular structure of nucleic acids. Nature 171, 737-738.
Anmerkung
1 Aufgrund der Degeneration des genetischen Codes werden dieselben Aminosäuren durch verschiedene Codons codiert. Die Bindung von Transkriptionsfaktoren an die DNA hat aber einen Einfluss auf die Wahl der verwendeten Codons für eine bestimmte Aminosäure.
Autor dieserNews: Harald Binder
© 2014, http://www.genesisnet.info/schoepfung_evolution/n207.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
05.12.12 Wieviel Evolution ist durch Kooption möglich?
Im „Zeitalter“ von Evo-Devo (evolutionäre Entwicklungsbiologie, https://www.genesisnet.info/index.php?Artikel=41266&Sprache=de&l=2) gilt für den evolutionären Wandel teilweise ein gegenüber dem Neodarwinismus neues Paradigma: Evolution erfolgt nicht nur durch Entstehung neuer Gene, sondern vor allem durch Änderungen der Nutzung und zunehmende Mehrfachnutzung vorhandener Gene. Man spricht in solchen Fällen von Kooptionen. Dies ist dadurch möglich, dass Änderungen in der Gen-Regulation erfolgen. Allerdings sind Vorgänge einer Kooption weitgehend hypothetisch. Eine Kooption konnte kürzlich durch eine ausgefeilte Studie wahrscheinlich gemacht werden (Rebeiz et al. 2011). Die Entstehung einer evolutionären Neuheit wird damit aber nicht gezeigt.
Gene an sich sind völlig inaktiv. Ihre Information kann nur genutzt werden, wenn aufgrund von Signalen eine Regulationskaskade ausgelöst wird, an deren Ende das Ablesen der Information des Gens steht (Genexpression). Unmittelbar vor dem Gen liegt der Promotor, an ihm muss die RNA-Polymerase* andocken, damit das Gen abgelesen und die entsprechende mRNA gebildet werden kann (= Transkription*), die die Vorlage für die Bildung eines entsprechenden Proteins ist.
*RNA-Polymerase (= Enzym, das die Synthese der messenger-RNA ermöglicht)
**Transkription (= „Übersetzung“ der DNA in mRNA)
Ebenfalls zur Gen-Regulation tragen sogenannte cis-Elemente bei. Dabei handelt es sich um kurze DNA-Bereiche, an denen (i. d. R. mehrere) Transkriptionsfaktoren* binden können, was entweder zur Verstärkung oder Verringerung der Polymerase-Aktivität und damit der Transkription führt. Ist die Wirkung verstärkend, werden sie als Enhancer bezeichnet. Cis-Elemente liegen meist vor dem Promotor, können aber auch weiter entfernt (sogar auf einem anderen Chromosom) oder im Gen selbst liegen.
*Transkriptionsfaktoren (= Proteine, die für den Start der Tätigkeit der RNA-Polymerase bei der Transkription von Bedeutung sind)
Vor diesem Hintergrund der Gen-Regulation kann Evolution nicht nur dadurch ablaufen, dass Gene selbst verändert werden, sondern auch durch Änderungen der Enhancer, also der DNA-Bereiche und derjenigen Proteine, die bei der Regulation der Genaktivität eine Rolle spielen. Änderungen in diesen Bereichen können dazu führen, dass Gene in anderer Weise als zuvor genutzt werden, das heißt, das Expressionsmuster (zeitlich oder Gewebe-spezifisch) der Gene ändert sich (zu „Genexpression“ s. o.). Über ein interessantes Beispiel einer solchen Änderung berichten Rebeiz et al. (2011). Ihnen gelang möglicherweise der Nachweis der Entstehung eines neuen Enhancers durch Kooption, d. h. ein ursprünglich vorhandener Enhancer wurde bei einem neuen Gen in veränderter Form neu genutzt.
Vier prinzipielle Möglichkeiten der Veränderung der Gen-Expression werden in der Literatur diskutiert (instruktive Abbildung unter http://ittakes30.wordpress.com/2011/05/31/something-new-under-the-sun).
- Transposition: Ein bereits existierender Enhancer gelangt an eine neue Stelle im Erbgut. Das führt zu einer neuen Ausprägung der Expression.
- Promotor-Switching: Es treten im Enhancer Änderungen in der Enhancer-Promotor-Spezifität auf, die dazu führen, dass ein anderer Promotor aktiviert wird.
- Kooption: Ein bereits existierendes genetisches Element wird in einem neuen Funktionszusammenhang verwendet. Das kann ein Gen für ein strukturelles Protein sein oder das können regulatorische Abschnitte sein, die neu verwendet werden
- Neuentstehung: Bisher nicht-funktionale DNA wird durch Mutationen zu einem Enhancer.
Rebeiz et al. (2011) konnten in ihren Untersuchungen die Kooption eines bereits vorhandenen Enhancers für eine neue Regulation der Genexpression in einem neuen Organ plausibel machen. Sie untersuchten 20 Gene einer Gruppe nah verwandter Taufliegen-Arten (Drosophila), um in jüngerer Zeit evolvierte neue Gen-Expressionsmuster zu untersuchen. Die Expression dieser Gene wurde in mehreren Larvenstadien der verschiedenen Drosophila-Arten ermittelt, und die Forscher entdeckten viele Unterschiede, die aber meistens geringfügig waren, z. B. Änderungen im zeitlichen Verlauf oder in der Größe der Körperregion, in der das Gen exprimiert wurde, oder solche, die auf Verlust zurückzuführen waren.
Ein Gen in der Art D. santomea aber hatte ein besonderes Expressionsmuster, das in den anderen Arten nicht vorkam. Es handelte sich um das Gen Neprilysin-1 (Nep1), das allgemein in den Flügeln, Beinen und im Zentralnervensystem aktiv ist. Nep1 kodiert eine extrazelluläre Metalloproteinase1 (Rebeiz et al. 2011, 10038). Bei D. santomea wird es, im Gegensatz zu allen anderen untersuchten Arten, zusätzlich auch in den sich entwickelnden Nervenzellen des entstehenden Sehlappens exprimiert. Die genaue Funktion von Nep1 in dem neuen Gewebe ist leider nicht bekannt. Die Untersuchung der Cis-Region von Nep1 offenbarte eine neue Enhancer-Aktivität in einer Region, die einige Mutationen angesammelt hatte. Dieser (neue) Enhancer überlappt mit anderen (bestehenden) Enhancer-Aktivitäten, von denen die neue Aktivität vermutlich kooptiert wurde.2 Weitere Experimente konnten ausschließen, dass es sich um ein altes Expressionsmuster handelt, das lediglich bei den anderen Arten verlorengegangen ist.
Die detaillierten Untersuchungen von Rebeiz et al. (2011) zeigen kurz zusammengefasst: Eine Änderung eines Enhancers des Gens Nep1 führt dazu, dass dieses Gen zusätzlich auch im Sehlappen exprimiert wird. Der zuvor schon vorhandene Enhancer bekommt somit eine weitere Funktion (Kooption). Diese wird in der weiteren Folge durch vier Mutationen optimiert.
Eine latente Aktivität war schon da. Die Autoren verglichen die Sequenzen des Enhancers von Nep-1 von D. santomea mit den Sequenzen nahe verwandter Drosophila-Arten, um die Sequenz des letzten gemeinsamen Vorfahren von D. santomea und D. yakuba zu rekonstruieren. Sie fanden heraus, dass der Enhancer des gemeinsamen Vorfahren eine signifikante Aktivität im Sehlappen besessen haben muss (etwa 40 % des Levels der Aktivität in D. santomea) und es auch bei entfernter verwandten Arten Spuren einer Aktivität gibt. Es liegt also bereits ein verborgenes Potential vor, das bei den meisten Drosophila-Arten latent bleibt, während es beim gemeinsamen Vorfahren von D. santomea und D. yakuba durch Mutationen zu einer moderaten Aktivität kam, die durch vier weitere Mutationen bei D. santomea zu einer Verstärkung der Expression führte. Bei D. yakuba führten weitere Mutationen zum Stilllegen der Expression.
Belegen die Befunde die Entstehung einer evolutionären Neuheit? Die Autoren versprechen sich von ihren Befunden Einblicke in die Entstehung von evolutionären Neuheiten (Rebeiz et al. 2001, 10041).3 Sie konnten zeigen, was auf molekularer Ebene im evolutionären Kontext passieren kann und haben dabei eine ausgezeichnete Arbeit abgeliefert, in der viele Aspekte berücksichtigt wurden, um die Schlussfolgerungen abzusichern.4 Aus ihren Schilderungen geht aber nicht hervor, ob die nachgewiesene Kooption eines Enhancers Auswirkungen auf den Phänotyp (= Erscheinungsbild) hat (in diesem Fall bei der Sehfähigkeit, da es um eine Änderung der Genexpression im Sehlappen handelt). Das betreffende Gen Nep1 kodiert eine extrazelluläre Metalloproteinase; das ist nicht neu. Neu ist, dass Nep1 auch im Sehlappen aktiv ist. Aber wie ist diese Aktivität einzuschätzen, welche Bedeutung hat sie? Die Kooption scheint nicht mit einer neuen Fähigkeit der Taufliegen einherzugehen. Es kann sich ohnehin bei einem einzigen Gen nur um einen (kleinen) Beitrag im komplexen System der Sehfunktion handeln. Möglicherweise hat die Kooption des Enhancers gar keine phänotypischen Auswirkungen.
Ob man von einer evolutionären Neuheit sprechen kann, hängt natürlich auch davon ab, was man unter „Neuheit“ (novelty) verstehen soll. Eine allgemein anerkannte Definition liegt nicht vor. Der Begriff scheint teilweise inflationär für alle möglichen Veränderungen verwendet zu werden. In dem von Rebeiz et al. untersuchten Fall bedeutet „Neuheit“ ein neues Expressionsmuster, das durch einige geringfügige, vielleicht vorher schon angelegte Änderungen zustande kam. Man kann das als „Neuheit“ bezeichnen, doch sind die dahinterstehenden Änderungen sehr geringfügig und erlauben keine weitergehenden Schlussfolgerungen.
Neue Genexpression oder latente Anlage? Vom evolutionsbiologischen Standpunkt aus könnte man dann in gewissem Sinne von einem Durchbruch sprechen, wenn eine Kooption eines Enhancers wirklich nachgewiesen wurde. Wie eingangs erwähnt werden Kooptionen theoretisch gefordert, aber ein Nachweis stattgefundener Kooptionen fehlt weitgehend. Die Wissenschaftler haben gezeigt, dass vier Mutationen ausreichen, um eine Genexpression an einem neuen Ort hochzuregulieren (daher handelt es sich um eine Kooption, s. o.). Allerdings diskutieren sie auch, dass diese Genexpression zuvor schon latent im Vorfahren angelegt war und auf geringem Level stattfand (s. o.), sie ist also nicht wirklich neu.5 Kann also wirklich von einer echten Kooption gesprochen werden?
Literatur
Rebeiz M, Jikomes N, Kassner VA & Carroll SB (2011) Evolutionary origin of a novel gene expression pattern through co-option of the latent activities of existing regulatory sequences. Proc. Natl. Scad. Sci. 108, 10036-10043 (doi: 10.1073/pnas.1105937108
Anmerkungen
1 Metalloproteinasen sind Enzyme, die die Peptidbindungen eines Proteins spalten können, wobei ein Molekül Wasser verbraucht wird und das Wassermolekül von einem oder zwei Metallkationen in Position gehalten wird. (nach Wikipedia)
2 „We traced the evolutionary history of the gain of Nep1 gene expression in the laminar neuroblasts of the optic lobe and found that the newly evolved Nep1 optic lobe enhancer was embedded within a region containing several enhancers that were active in other tissues. Furthermore, we demonstrated that the optic lobe enhancer included sequences required for the activity of other enhancers, which indicated that the enhancer activity evolved by co-option of preexisting, long-conserved, regulatory sequences. Finally, we revealed that mutations at four sites elevated the latent low optic lobe activity of the ancestral regulatory sequence to the present-day high level in D. santomea“ (S. 10041).
3 „These results highlight the ease with which evolution may “tinker” with regulatory sequences to generate novelty…“ (S. 10041)
4 Unter anderem die Untersuchung mehrerer Entwicklungsstadien, um auszuschließen, dass man Genexpressionen nur übersehen hat, weil man zur falschen Zeit geguckt hat, und eine Reihe von Tests, um alternative Erklärungsmöglichkeiten auszuschließen.
5 „We suggest that the novel optic lobe enhancer evolved by exploiting the cryptic activity of extant regulatory sequences, and this may reflect a general mechanism whereby new enhancers evolve.“
Autor dieserNews: Reinhard Junker
© 2012, http://www.genesisnet.info/schoepfung_evolution/n189.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
09.02.12 Am Anfang die Vielfalt?
Die sehr artenreiche Ameisengattung Pheidole bildet gewöhnlich zwei Arbeiterkasten aus, die unterschiedlich groß sind und in den Nestern verschiedene Aufgaben erfüllen. Bei einigen wenigen Arten gibt es darüber hinaus eine sehr große Kaste von „Supersoldatinnen“ mit extrem großen Köpfen. Bisher war angenommen worden, dass sie sich unabhängig entwickelt haben. Doch einige neue Befunde sprechen nun dafür, dass das genetische Potential für die Supersoldatinnen schon im gemeinsamen Vorläufer der Gattung Pheidole vorhanden war und auch heute noch in vielen Arten abrufbar ist. Dieser Befund passt gut zum Konzept der polyvalenten Grundtypen.
„Zeig, was in Dir steckt!“ Wer diese Aufforderung ausspricht, rechnet damit, dass bei einer Person mehr Potential vorhanden ist als aktuell sichtbar. Immer wieder zeigt sich das auch in der Tier- und Pflanzenwelt. Gut erforschte Beispiele sind Panzerung und Bauchstacheln bei Stichlingen oder Flügel bei Stabheuschrecken, die optional ausgebildet werden können (in verschiedenen Variationen oder auch gar nicht). Im Erbgut schlummern manche Möglichkeiten, die fast wie auf Knopfdruck abgerufen werden können. Im Zusammenhang mit der Debatte um Schöpfung und Evolution interessiert die Frage, woher dieses Potential kommt und ob dessen Herkunft durch naturwissenschaftliche Forschung geklärt werden kann. Nach dem Grundtypmodell der Schöpfungslehre sind die Grundtypen (die als Schöpfungseinheiten interpretiert werden können) ursprünglich polyvalent, das bedeutet unter anderem: sie sind mit verschiedenen genetischen und phänotypischen (gestaltlichen) Möglichkeiten ausgestattet, die teilweise nur bei Bedarf aktiviert und abgerufen werden können (vgl. Artikel Genetisch polyvalente Stammformen von Grundtypen, https://www.genesisnet.info/index.php?Artikel=1244&Sprache=de&l=1). Von einem Beispiel, das diese Vorstellungen (und damit das Grundtypkonzept) unterstützt, berichten Forscher um Ehab Abouheif von der kanadischen McGill Universität (Rajakumar et al. 2012). Auch wenn diese Forscher ihre Beobachtungen und Resultate nicht im Zusammenhang mit dem Grundtypmodell diskutieren, liest sich ihr Artikel über die gelegentliche Ausbildung von „Supersoldatinnen“, einer besonderen Kaste bei verschiedenen Ameisenarten, fast wie ein Kapitel über Grundtypenbiologie und polyvalente Stammformen.
Die Forscher wollten herausfinden, wie neue Arbeiterkasten bei Ameisen entstehen. Dazu untersuchten sie Ameisen der mit über 1000 Arten sehr artenreichen und weltweit verbreiteten Gattung Pheidole. Diese Ameisen gehören zur Unterfamilie der Knotenameisen (Myrmicinae). Ihre Arbeiterkaste ist stark größendimorph, das heißt es gibt zwei Typen von unfruchtbaren Weibchen: Kleinere, 2-4 mm große Arbeiterinnen mit gewöhnlichem Körperbau – sie kümmern sich um das Nest und um das Futter – und größere Arbeiterinnen, die auch als Soldatinnen bezeichnet werden und im Vergleich zu ihrem Körper sehr große Köpfe und „Beißzangen“ (Mandibeln) besitzen. Die Soldatinnen verteidigen das Nest und verarbeiten das Futter.
Darüber hinaus sind aus den Wüsten Nordamerikas bei acht Pheidole-Arten aber auch noch sogenannte Supersoldatinnen bekannt. Sie sind mehr als doppelt so groß wie normale Soldatinnen und haben riesige Köpfe und Kiefer, mit denen sie die Eingänge zum Nest blockieren, wenn sie von Wanderameisen angegriffen werden, denen die normalen Soldatinnen nicht gewachsen sind. Andere Aufgaben erfüllen sie kaum, sie müssen sogar gefüttert werden.
Kürzlich erlebten die Forscher eine Überraschung: Auch auf Long Island entdeckten sie bei der dort ansässigen Ameisenart Pheidole morrisi Soldatinnen, die riesige Köpfe besaßen. Bei dieser Art waren Supersoldatinnen zuvor noch nie beobachtet worden. Es wurde auch nicht damit gerechnet, denn auf Long Island gibt es keine Wanderameisen. Daraufhin untersuchten sie, ob auch andere Pheidole-Arten im Labor Supersoldatinnen hervorbringen können. Und das funktionierte nicht nur bei Pheidole morrisi, sondern auch bei zwei weiteren Arten tatsächlich: Das Supersoldatinnen-Potential kann während des Larvenstadiums aktiviert werden, wenn zum richtigen Zeitpunkt die richtige Konzentration des Juvenilhormons (bzw. eines chemischen Ersatzstoffes) und passende Nahrung verabreicht wird. Das Juvenilhormon verzögert die Umwandlung (Metamorphose) der Larve zum erwachsenen Stadium, so dass die Chitinhaut länger wachsen kann. Nun vermuten die Forscher, dass sogar alle Pheidole-Arten diese Möglichkeit besitzen, Supersoldatinnen heranwachsen zu lassen, die bei Auftreten vieler Feinde aktiviert werden kann.
Es war schon vorher bekannt, dass nicht alle der bisher bekannten acht Pheidole-Arten, die Supersoldatinnen ausbilden, näher miteinander verwandt sind. Daher war angenommen worden, dass diese Kaste zweimal unabhängig evolutionär entwickelt wurde. Aufgrund der neuen Befunde bezweifelt die Gruppe um Abouheif diese Deutung. Wenn durch Hormongabe die Super-Kaste erzeugt werden kann, müssen die genetischen Voraussetzungen dafür schon im Erbgut stecken. Die Ausbildung von Supersoldatinnen gelang auf diese Weise bei Arten aus ganz verschiedenen Positionen im Pheidole-Stammbaum. Aufgrund der weiten Verteilung dieses Potential liegt die Annahme nahe, dass es ursprünglich allen Pheidole-Arten zu eigen war. Seine Ausprägung scheint nur vom richtigen Timing und der passenden Menge Juvenilhormon und der Ernährung abzuhängen. Dass die Super-Kaste nur bei besonderem Bedarf ausgebildet wird, ist insofern verständlich, als ihre Produktion viel Energie kostet. Rajakumar et al. (2012, 81) schreiben: „Das Entwicklungspotential für die Produktion der Supersoldatinnen wurde beibehalten und war wahrscheinlich im gemeinsamen Vorfahren alle Pheidole-Arten vorhanden. Ohne das Wissen über dieses ursprüngliche Entwicklungspotential hätten wir angenommen, dass die Supersoldatinnen-Kaste neu evolviert sei.“ Wahrscheinlicher sei aber, so führen sie weiter aus, dass die Ausbildung der Supersoldatinnen-Kaste bei den meisten Arten verlorengegangen sei, während das Potential bei einigen Arten dafür erhalten blieb. Der Schlusssatz der Autoren, dass durch solche Prozesse die Evolution „neuer“ Phänotypen (im Sinne von de novo) erleichtert werde, ist durch die von ihnen berichteten Ergebnisse daher nicht begründet.
Der Vorgang der Aktivierung bzw. Reaktivierung der Supersoldatinnen ist ein Beispiel für „genetische Akkommodation“ (Rajakumar et al. 2012, 81). Darunter versteht man die Ausweitung der phänotypischen Formenvielfalt aufgrund äußerer (Extrem-)Einflüsse (hier die Entstehung der Supersoldatinnen-Kaste, z. B. durch Konfrontation mit Wanderameisen) und die nachfolgende Beibehaltung des neuen (reaktivierten) Phänotyps durch Selektion der Gene, die die Häufigkeit seiner Ausprägung positiv beeinflussen (hier Überleben der Kolonie nach einem Angriff) (vgl. Artikel Evo-Devo, https://www.genesisnet.info/index.php?Artikel=41266&Sprache=de&l=2).
Bemerkenswert ist, dass trotz des Fehlens der Supersoldatinnen bei gewöhnlich 99% der Pheidole-Arten das Potential für diese Kaste nicht im Laufe der Zeit verlorengegangen ist, sondern aktiviert werden kann. Im Allgemeinen wird nicht genutzte Erbinformation durch Verlustmutationen im Laufe der Zeit verloren gehen. Nach phylogenetischen Analysen sollen sich die Gattung Pheidole schon vor 30 bis 65 Millionen Jahren in die rund 1100 verschiedenen Arten auseinanderentwickelt haben. Warum ist das Potential für die Super-Kaste dennoch in verschiedenen Arten vorhanden? Die Forscher vermuten, dass Supersoldatinnen-Potential mitsamt seinen Genen und ontogenetischen Entwicklungswegen nicht beseitigt werden kann, weil es auch bei der Bildung der normalen Soldatinnen benötigt wird (Rajakumar et al. 2012, 82).
Abbildung
Literatur
Rajakumar R, San Mauro D, Dijkstra MB, Huang MH, Wheeler DE, Hiou-Tim F, Khila A, Cournoyea M & Abouheif E (2012) Ancestral Developmental Potential Facilitates Parallel Evolution in Ants. Science 335, 79-82.
Autor dieserNews: Reinhard Junker
© 2012, http://www.genesisnet.info/schoepfung_evolution/n181.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
21.06.11 Die Entstehung neuer Enzyme – ganz einfach?
Die evolutionäre Entstehung eines Enzyms mit neuen funktionellen Eigenschaften im pflanzlichen Sekundärstoffwechsel aus einem vorhandenen Enzym des Primärstoffwechsels konnte durch experimentelle und vergleichend-biologische Untersuchungen plausibel gemacht werden. Die experimentell erfolgten Einzelschritt-Änderungen von Aminosäurebausteinen waren jede für sich selektionspositiv. Die hier vorgestellte Entstehung neuer Enzymfunktionen ist jedoch gemessen an den bisher bekannten Variationsmechanismen nicht überraschend und kann als Ausdruck einer bereits angelegten Polyvalenz im Wechselspiel von Genen und der durch sie codierten Proteinen interpretiert werden.
Pflanzen sind gegen Fraß nicht wehrlos: So nutzen viele Kreuzblütler wie das Wiesenschaumkraut (Cardamine pratensis) oder die viel untersuchte Acker-Schmalwand (Arabidopsis thaliana) Senfölglykoside, um sich gegen Fressfeinde (z. B. Raupen) zur Wehr zu setzen. Diese „Senfölbomben“ sind sekundäre Stoffwechselprodukte und werden bei Beschädigung der Pflanze zur Abwehr gegen den Schädling freigesetzt. Forscher des Max-Planck-Instituts für chemische Ökologie in Jena haben nun eine interessante Entdeckung gemacht: Ein Enzym, das für die Bildung der Senfölglykoside benötigt wird – Methylthioalkylmalat-Synthase (MAM) – ist in seiner Struktur dem Stoffwechselenzym Isopropyl-Malat-Synthase (IPMS) sehr ähnlich (de Kraker & Gershenzon 2011). Die IPMS hat aber eine ganz andere Funktion, sie ist bei der Bildung der Aminosäure Leucin beteiligt und damit Teil des Primär-Stoffwechsels.
Die beiden Enzyme unterscheiden sich in nur zwei bedeutsamen strukturellen Merkmalen: Bei MAM fehlen im Vergleich zur IPMS die letzten 120 Aminosäuren und in seinem aktiven Zentrum sind zwei Aminosäuren ausgetauscht. (Weitere vorhandene Unterschiede haben offenbar keinen Einfluss auf die Struktur und die Funktion der Enzyme.) Das bei der MAM fehlende letzte Stück hat bei der IPMS die Funktion einer Rückkopplung: Ist genügend Leucin vorhanden, wird die Produktion der IPMS gedrosselt. Fehlt diese 120-Aminosäure-Kette, geht die Produktion unvermindert weiter und kann nicht mehr kontrolliert werden. Das Fehlen dieser Kette hat weiter zur Folge, dass sich das verkürzte IPMS-Enzym anders faltet; die Architektur (Quartärstruktur) ist anders als zuvor. Die Verkürzung der Kette wirkt sich auch auf das aktive Zentrum aus, so dass das verkürzte IPMS größere Moleküle binden kann und nun die Bildung von Vorstufen der Senfölglykolsiden anstelle der Bildung von Leucin unterstützt.
Aufgrund dieser Beobachtungen schlagen de Kraker & Gershenzom (2011) folgendes evolutionäre Szenario vor: Ausgangsstoff für die Entstehung der MAM, also des Enzyms, das bei der Bildung der Senfölglykoside benötigt wird, ist die um 120 Aminosäuren längere IPMS. Dessen codierendes Gen wurde zunächst verdoppelt (Gen-Duplikation). Anschließend ging beim Duplikat die 120-Aminosäuren-Kette verloren, was die oben beschriebenen Auswirkungen zur Folge hatte. Die dadurch veränderte Substrataffinität zu größeren Molekülen wurde weiter durch Punktmutationen, die das aktive Zentrum trafen, optimiert, so dass das heute vorliegende MAM entstand.
De Kraker & Gershenzom (2011) haben dieses hypothetische Szenario in Laborexperimenten nachgestellt und die oben beschriebene Abfolge simuliert. Somit konnten sie zeigen, dass dieser Weg in der Geschichte der Kreuzblütler-artigen Pflanzen (Ordnung Brassicales) so abgelaufen sein könnte. Dies sei ein Beispiel dafür, dass ein Enzym aus dem Primärstoffwechsel für einen anderen Zweck im Sekundärstoffwechsel rekrutiert (sozusagen „zweckentfremdet“) wurde, so die Autoren.
Welche Schlussfolgerungen können aus diesen Beobachtungen und Experimenten gezogen werden? Zunächst legt diese Untersuchung nahe, dass Enzyme mit neuen Eigenschaften bzw. Fähigkeiten ohne gezielten Eingriff entstehen können. (Die experimentelle Rekonstruktion wurde geplant und geschah gezielt, macht aber plausibel, dass ein evolutiver Vorgang so abgelaufen sein könnte.) Die Entstehung der neuen Funktion kann über Einzelschritte (einzelne Mutationen) nachvollzogen werden, die jeweils selektierbar waren. Es muss nicht angenommen werden, dass mehr als eine Mutation gleichzeitig auftreten musste, um zum nächsten selektierbaren Zustand zu gelangen.
Im Vergleich zur bisher bekannten Variationsfähigkeit der Lebewesen und deren Grenzen ist dieser Vorgang nicht überraschend (vgl. Behe 2007). Erstaunlich ist aber, dass durch relativ einfache Änderungen – Genduplikation, Verlust, Punktmutation – aus einem Enzym des Primärstoffwechsels ein Enzym des Sekundärstoffwechsels mit einer grundsätzlich anderen Funktion werden kann. Es ist festzuhalten, dass der Ausgangspunkt ein funktionales Enzym war und die neue Funktion im Wesentlichen auf einen Verlust zurückzuführen ist (die nachfolgenden Punktmutationen im aktiven Zentrum führten lediglich zu dessen Optimierung).
Welche Schlussfolgerungen können nicht gezogen werden? Inwieweit dieses Beispiel verallgemeinert werden kann, müssen weitere Beispiele zeigen. Junker & Scherer (2006, 144f.) schildern schon länger bekannte Beispiele der Entstehung einer neuen Substratspezifität, bei denen allerdings die neue Funktion relativ ähnlich wie die bisherige bleibt.
Eine Verallgemeinerung und Übertragung der von De Kraker & Gershenzom vorgeschlagenen Mechanismen auf die Entstehung anderer Enzyme ist nur möglich, wenn es sich um Änderungen handelt, bei denen jeder einzelne Schritt selektierbar ist. Die Studie kann die generelle Frage jedoch nicht beantworten, ob auf evolutivem Wege Änderungen von Enzymstrukturen und ihren Funktionen möglich sind, die mehr als einen Schritt benötigen, um von einem selektierbaren Zustand zum nächsten zu gelangen.
Ob also die Entstehung neuer Enzyme „oft verblüffend einfach“ ist, wie die AG Evolutionsbiologe eine Newsmeldung überschreibt, muss sich erst noch zeigen (vgl. http://ag-evolutionsbiologie.de/app/download/4566099102/kraker-gershenzon-2011.html). Schwer zu beurteilen und auf der Basis naturwissenschaftlicher Argumentation vorerst nicht entscheidbar bleibt die Frage, ob es sich bei den dargestellten strukturell-funktionellen Änderungen um einen evolutionär-glücklichen Zufallstreffer oder um eine vorprogrammierte Situation handelt. Letztere wäre dann Ausdruck einer angelegten Polyvalenz im Wechselspiel von Genen und der durch sie codierten Proteine im Netzwerk der globalen zellulären Stoffwechselprozesse. Die vorgestellten Befunde erlauben beide Interpretationsmöglichkeiten.
Literatur
Behe M (2007) Edge of Evolution. The search for limits of Darwinism. Free Press, New York.
De Kraker JW & Gershenzon J (2011) From Amino Acid to Glucosinolate Biosynthesis: Protein Sequence Changes in the Evolution of Methylthioalkylmalate Synthase in Arabidopsis. The Plant Cell 23, 38-53.
Junker R & Scherer S (2006) Evolution – ein kritisches Lehrbuch. Gießen.
Autor dieserNews: Reinhard Junker
© 2011, http://www.genesisnet.info/schoepfung_evolution/n170.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
20.04.11 Von komplex nach einfach?
Evolution bedeutet den schrittweisen Aufbau der Baupläne der Lebewesen. Immer wieder aber muss aufgrund von Merkmalsvergleichen angenommen werden, dass Entwicklungen in die umgekehrte Richtung abgelaufen sein müssten, wenn eine Evolution abgelaufen wäre, und dass hypothetische Stammformen von Tiergruppen komplex gewesen sein müssen. Woher aber kommen diese hypothetischen Vorläufergruppen? Unklar ist auch, weshalb in evolutionstheoretischer Sicht Organe immer wieder verloren gegangen sind.
Vor zwei Jahren erhielt ein unscheinbares Geschöpf besondere Aufmerksamkeit: Der zu den Placozoa („Plattentiere“) gehörende sehr einfach gebaute Vielzeller Trichoplax adhaerens, der als „Urvater“ der Tiere galt, wurde überraschend aufgrund umfangreicher und vielseitiger Untersuchungen in eine abgeleitete phylogenetische Position umgruppiert (zu Einzelheiten siehe Der einfachste Vielzeller kommt groß heraus, https://www.genesisnet.info/index.php?News=166).
Die Folge davon ist, dass angenommen werden muss, dass der gemeinsame hypothetische Vorfahr von Trichoplax adhaerens und der anderen Vielzeller überraschend komplex war. Einer der Bearbeiter, Prof. Schierwater, äußerte, dass aufgrund der Ergebnisse eine unerwartete Parallelentwicklung von niederen und höheren Tieren angenommen werden müsse und dass in Frage gestellt sei, dass sich komplexere Formen graduell aus einfacheren Formen ableiten. „Hier müssen wir wohl umdenken“ (http://idw-online.de/pages/de/news297821). Ein genetisch komplexer Vorfahr wurde schon seit einiger Zeit angenommen, weil sich gezeigt hat, dass bei verschiedensten Tiergruppen die gleichen Regulationsgene, die für die Bildung von Organen erforderlich sind, aktiv sind. Diese müssten laut evolutionstheoretischer Argumentation auch im hypothetischen gemeinsamen Vorfahren präsent gewesen sein (vgl. Evo-Devo, https://www.genesisnet.info/index.php?Artikel=41266&Sprache=de&l=2).
Zwei neue Studien anderer Tiergruppen weisen nun erneut in diese Richtung: Evolutionäre Stammformen müssen als sehr komplexe Lebensformen betrachtet werden.
Acoela. Zunächst soll über eine Studie an winzigen Würmern berichtet werden. Es handelt sich um die Gruppe der Acoela, deren Name vom Fehlen eines Coeloms (Körperhohlraum) abgeleitet ist. Die Acoela sind stark abgeflachte Würmer, sind meist unter 2 Millimeter lang, haben nur eine einzige Körperöffnung und besitzen keinen Magen. Den Besitz von nur einer einzigen Körperöffnung haben sie mit den Cnidaria (Seeanemonen und Quallen gemeinsam, anders als diese haben sie drei und nicht nur zwei embryonal angelegte Gewebeschichten (Keimblätter), weshalb sie als willkommene Zwischenformen zwischen den Cnidariern und höheren Tieren galten. Eine neue Studie von Telford und Mitarbeitern (Philippe et al. 2011) kommt zu dem Ergebnis, dass die Acoela an die Basis der Deuterostomier (Neumundtiere) in die Nähe der Stachelhäuter (zu denen beispielsweise Seeigel gehören) zu stellen sind, ein deutlicher Sprung nach oben im hypothetischen phylogenetischen Stammbaum. Ihre genetischen Analysen legen nahe, dass die Acoela und der marine (= im Meer lebende) Wurm Xenoturbella von einer komplexeren Form abstammen muss und die meisten Merkmale, die andere Deuterostomier auszeichnen, während ihrer Stammesgeschichte offensichtlich verloren haben (vgl. Maxmen 2011). Zu dieser Schlussfolgerung gelangten die Autoren aufgrund der Auswertung von drei verschiedenen unabhängigen Datensätzen (Kern-DNA, mitochondriale DNA und microRNA). Maxmen (2011, 162) kommentiert: „Wenn die Acoela zu den Deuterostomiern passen, müssen diese Würmer von einem Vorfahren mit einem Zentralnervensystem, einer Körperhöhle und einem durchgehenden Verdauungssystem, das Mund und Anus verbindet, evolviert sein – Merkmale, die bei den heute lebenden Deuterostomiern anzutreffen sind. Daher müssen die Forscher erklären, wie die Acoela und Xenoturbella diese und andere Merkmale verloren haben. Sie müssen auch nach einer anderen primitiven Linie Ausschau halten, die den evolutionären Schritt zwischen quallenartigen Tieren und den Bilateriern (Zweiseitentiere) repräsentieren.“
Die neue Stellung der Acoela ist allerdings noch umstritten. Sollte sie aber zutreffen, gilt einmal mehr, dass im evolutionstheoretischen Rahmen Entwicklungen von komplex zu einfach angenommen werden müssen und eine vorhandene Lücke zwischen verschiedenen Tiergruppen sich noch größer darstellt als zuvor angenommen.
Ringelwürmer. Die zweite Studie, über die hier kurz berichtet werden soll, handelt von den Ringelwürmern (Annelida), deren interne Beziehungen in einer phylogenomischen Analyse untersucht wurden (Struck et al. 2011). Dabei wurden 34 Anneliden-Taxa und 47.953 Aminosäure-Positionen von 231 Proteinen in die Untersuchungen einbezogen. In der Geschichte der Systematik dieser Gruppe gab es manche Umgruppierungen, die Struck et al. in einem kurzen Überblick erläutern. Die Wissenschaftler erzielten nun ein „überraschend klares Ergebnis“ (Arendt 2011). Demnach ist die Hauptgruppe der Anneliden deutlich unterteilt in die beiden Gruppen Errantia (frei bewegliche Arten) und Sedentaria (teilweise festsitzende grabende oder in Röhren lebende Arten). Auch hier folgt aus der neuen Phylogenie, dass angenommen werden muss, dass der letzte gemeinsame Vorfahre der Anneliden ein mit Sinnesorganen „reichlich ausgestattetes“ Tier gewesen sein muss (Arendt 2011). Legt man die morphologischen (gestaltlichen) Merkmale zugrunde, gibt es ein „schwerwiegendes Problem“: die offenkundige Leichtigkeit, mit der Merkmale während der Evolution verloren gehen können. Arendt illustriert dies am Beispiel der Palpata, einer Anneliden-Gruppe deren Mitglieder durch den Besitz spezieller Kopf-Anhänge, sogenannten Palpen, gekennzeichnet sind. Aufgrund der neuen Anneliden-Phylogenie muss ein unabhängiger Verlust von Palpen bei den Errantia und den Sedentaria angenommen werden. „Dieses Beispiel bestätigt die allgemeine Idee des häufigen und unabhängigen Verlusts von Merkmalen während der Evolution der Tiere“, kommentiert Arendt (2011, 44).
Befunde wie diese werfen zum einen aber immer wieder neu die Frage auf, wie die komplexeren Vorfahren entwickeln konnten. Die Merkmale, die verloren gegangen sein sollen, müssen schließlich zuvor erst einmal entstanden sein. Außerdem muss es Ursachen geben, die zum Verlust geführt haben. Zum anderen stellen sich die unerwarteten Merkmalsverteilungen in einer nicht-evolutionären Perspektive in einem anderen Licht dar: Wenn Merkmale im Prinzip frei kombinierbar sind, sind „unpassende“ Merkmalskonstellationen verständlich und die Frage nach den Gründen eines Verlustes stellt sich nicht.
Arendt (2011) weist in seinem Kommentar auch darauf hin, dass die methodische Stärke der Cladistik gleichzeitig große Probleme erzeugt: eine Inkonsistenz von Ergebnissen von verschiedenen methodischen Ansätzen der Phylogeneseforschung. Die molekularen Verwandtschaften führen schon fast regelmäßig zu Umgruppierungen, die auf der morphologischen Ebene die Annahme erzwingen, dass Vorfahren (relativ) komplex waren und Merkmal häufig verloren gingen.1 Wenn verschiedene Methoden immer wieder zu widersprüchlichen Ergebnissen führen, könnte dies ein Indiz auf ein grundsätzliches nicht erkanntes Problem sein.
Literatur
Arendt D (2011) Annelid who’s who. Nature 471, 44-45.
Maxmen A (2011) A can of worms. Nature 470, 161-162.
Philippe H et al. (2011) Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470, 255-258.
Struck TH, Paul C, Hill N, Hartmann S, Hösel C, Kube M, Lieb B, Meyer A, Tiedemann R, Purschke G & Bleidorn C (2011) Phylogenomic analyses unravel annelid evolution. Nature 471, 95-99.
Anmerkung
1 „The case of the annelids exemplifies both the beauty and the pitfalls of phylogeny reconstruction when applying the principle of parsimony, which settles on the tree minimizing gain or loss of particular characteristics. At the molecular level, this approach has proved very powerful, and it has been further enhanced by the advent of phylogenomics. But it is becoming increasingly obvious that, on the basis of morphological characteristics alone, there is a serious problem: the apparent ease with which such characteristics are lost“ (Arendt 2011).
Autor dieserNews: Reinhard Junker
© 2011, http://www.genesisnet.info/schoepfung_evolution/n166.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
17.01.11 Dunkle Seiten des Genoms beleuchtet
Waren bis vor wenigen Jahren fast ausschließlich die proteincodierenden Gene des Erbguts im Fokus der Forschung, so weisen Erkenntnisse aus den vergangenen zehn Jahren auf unterbelichtete Bereiche hin. Im Erbgut ist viel mehr als nur die Anleitung für Eiweißsynthese zu finden und man darf gespannt sein, was unter neuem Licht noch alles erkennbar werden wird.
Das Lehrbuchwissen war zunächst einfach und übersichtlich: Die DNA im Genom (= Erbgut) enthält die Anweisung zum Aufbau von Proteinen. Die Anleitungen sind in Gene unterteilt und werden durch chemisch verwandte Nukleinsäuren, die RNA, aus dem Zellkern an den Ort der Proteinsynthese, die Ribosomen gebracht. Zwischen den einzelnen Genen liegen im Genom weite Bereiche, denen keine Funktion und Bedeutung zugeordnet werden konnte („junk DNA“, „genetischer Abfall“).
Forschung der vergangenen zehn Jahre hat zunehmend Licht auf die umfangreichen zunächst als nutzlos erscheinenden Bereiche des Genoms geworfen. Die Wissenschaftsjournalistin Elizabeth Pennisi (2010) hat für die Zeitschrift Science bedeutende Veröffentlichungen aus diesem Zeitraum kurz zusammengefasst.
Nach derzeitigem Kenntnisstand sind im menschlichen Genom rund 21.000 Gene enthalten, die für Proteine codieren. Sie nehmen ca. 1,5 % des Genoms in Anspruch. Schon früher wurde bezweifelt, dass der Rest – immerhin mehr als 98% – nutzloser Abfall sei, da der Ballast in diesem Umfang gerade auch nach evolutionären Vorstellungen im Laufe der Zeit hätte entsorgt werden müssen.1
Pennisi führt folgende Befunde an, die das Genom in einem neuen Licht erscheinen lassen:
- Der Mensch weist nicht nur viele proteincodierende Gene auf, die sich auch im Genom der Maus finden, sondern auch weite Teile der nicht-codierenden DNA sind in beiden Organismen ähnlich. Ohne Funktion dieser Bereiche ist der Befund nur schwer erklärbar, da bei Funktionslosigkeit Mutationen nicht durch Selektion korrigiert werden und sich so anhäufen. Die entsprechenden DNA-Sequenzen in verschiedenen Organismen werden dadurch immer unähnlicher. Untersuchungen an Mäuseembryonen belegen, dass nicht codierende Bereiche des Genoms eine Rolle in der Genregulation spielen. Manche Gene, deren Aktivität sie steuern, liegen im Genom weit von ihnen entfernt.
- Auch andere umfangreiche Studien, in denen genetische Risikofaktoren für Krankheiten untersucht wurden, lieferten Hinweise auf Funktionalität von nicht codierenden Genombereichen: ca. 40 % von DNA-Sequenzen, in denen sich gesunde und erkrankte Individuen um eine einzige DNA-Base unterscheiden, liegen in Bereichen zwischen den Genen.
- In anderen Arbeiten wurde gezeigt, dass weitaus mehr DNA in RNA umgeschrieben wird als nur für die Boten-RNA (mRNA) oder die RNA der Ribosomen (rRNA). Etwa 80 % der DNA einer Zelle scheinen in RNA umgeschrieben zu werden, ohne dass vom größten Teil dieser transkribierten Bereiche bisher bekannt ist, was deren Funktion ist.
- Bei Forschungen an Pflanzen und Fadenwürmern wurden Mechanismen entdeckt, wie man mit kleinen RNA-Fragmenten Gene ausschalten kann. Die Erforschung und Entwicklung der als RNA-Interferenz (RNAi) bezeichneten Methode wurde 2006 mit einem Nobelpreis ausgezeichnet. In vielen Untersuchungen wurde bestätigt, dass sehr kurze RNA-Fragmente aus 21 bis 30 Basen mit den Chromosomen wechselwirken und so die Aktivität von Genen kontrollieren und steuern können. Wenn in Hefezellen einzelne dieser kurzen RNA-Sequenzen fehlen, ist die Zellteilung gestört, und auch an Entwicklungsprozessen und Krebsentstehung sind sie beteiligt.
- Aber nicht nur diese kleinen RNA-Schnipsel erregten Aufsehen, auch die so genannte große zwischen den Genen liegende, nicht codierende RNA (large intervening noncoding RNA, lincRNA), entfaltet regulatorische Funktion auf Gene. Es könnte sein, zumindest äußern sich manche Forscher in diese Richtung, dass dieser Anteil des Genoms sich in seiner Bedeutung als ebenso bedeutsam erweist wie die proteincodierenden Gene.
Abschließend stellt Pennisi fest, dass vor zehn Jahren vom Genom fast ausschließlich die proteincodierenden Gene im Blickfeld der Forschung waren. Heute gilt das Interesse auch vielen anderen Bereichen des Erbguts. Ihre zusammenfassende Darstellung kann Staunen auslösen über die faszinierende Vielfalt und Dichte an Steuer- und Regulationsmechanismen in Zellen.
Der Rückblick der Autorin dokumentiert aber gleichzeitig, dass durch die Betrachtungsweise des Forschungsgegenstands – oder um ihr Bild aufzugreifen; seine Beleuchtung – auch Schatten produzieren: einzelne Aspekte werden betont, andere vernachlässigt oder gar ausgeblendet.
Die aktuelle Erforschung des Genoms zeigt, dass bei allen faszinierenden Erkenntnissen sich viele Hoffnungen nicht erfüllt haben. Der Blickwinkel muss erweitert werden, denn das Genom an sich liefert nicht die erhofften entscheidenden Einblicke in die grundlegenden Ursachen des Lebens, sondern es erscheint zunehmend als Bestandteil eines umfassenden und komplexeren Systems. Wenn bisher dunkle Bereiche vom Lichtkegel neuer Fragestellungen erfasst werden, bewirkt das einerseits Ernüchterung hinsichtlich erhoffter entscheidender Antworten aus dem Genom; gleichzeitig werden aber auch neue Horizonte für spannende Forschung in einem neuen Licht eröffnet.2
Literatur
Pennisi E (2010) Shining a light on the genome’s ‚Dark Matter‘. Science 330, 1614.
Anmerkungen
1 Dennoch wurde und wird die mutmaßliche „junk DNA“ als Argument gegen die Vorstellung von optimalem Design ins Feld geführt.
2Angesichts des Forschungsstands über die „Dark Matter“ des Genoms sollte die Vorstellungen von der „Junk-DNA“ vorerst ad acta gelegt werden. Das mit der „junk DNA“ begründete Argument, es gebe Design-Fehler und das spreche gegen eine Schöpfung, ist daher auch sehr fragwürdig geworden.
Autor dieserNews: Harald Binder
© 2011, http://www.genesisnet.info/schoepfung_evolution/n160.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
08.01.11 Langzeit-Experiment mit Drosophila – weniger Evolution als gedacht
Um genetische Veränderungen im Verlauf vieler Generationen experimentell zu untersuchen ist man auf Organismen angewiesen, die eine kurze Generationszeit aufweisen. Entsprechende Langzeitstudien mit Bakterien und Hefekulturen sind beschrieben. Diese sich asexuell fortpflanzenden Organismen stellen die bisherige empirische Basis dar für Modelle und Mechanismen von Langzeit-Evolutionsprozessen.
Burke et al. (2010) stellten kürzlich eine Studie mit der Taufliege Drosophila melanogaster vor – einem klassischen „Haustier“ der Genetiker. Damit liegt erstmals eine umfangreiche genetische Studie eines Langzeit-Evolutionsexperiments mit Organismen vor, die sich sexuell fortpflanzen.
Im Labor von M. Rose werden die kleinen Zweiflügler seit 1991 inzwischen in mehr als 600 Generationen gezüchtet und auf schnellere Entwicklung selektiert. Die gezüchteten Populationen entwickeln sich im Verhältnis zu Tieren der Ausgangs- und Vergleichsgruppe ca. 20 % schneller vom Ei bis zum erwachsenen, fortpflanzungsfähigen Tier. Damit einher geht auch die Entstehung veränderter Phänotypen* (etwa bezüglich Größe, Lebensdauer etc.).
*Phänotyp (= äußeres Erscheinungsbild)
Für die Untersuchung wurden Daten aus umfangreichen Genomanalysen erzeugt. Es wurden sowohl spezielle Gene analysiert (bezüglich Änderung in der Allelhäufigkeit: allele frequency differentiation) als auch komplette Genomdaten von Drosophila-Populationen aus dem Selektionsexperiment herangezogen. (Allele sind Varianten eines Gens.)
Bisher war man meist davon ausgegangen, dass bei sexueller Fortpflanzung wie auch bei der Entwicklung von Bakterienkulturen genetische Veränderungen (Mutationen) in einer Population auftauchen und dann in einem bestimmten Erbgutabschnitt fixiert werden. In ihrer Arbeit suchten Burke et al. nach positiven (Punkt-)Mutationen, die ein neues Basenpaar und damit einen neuen sog. SNP (Einzelnukleotid-Polymorphismus) erzeugen. Von Polymorphismus spricht man, wenn ein Gen (bzw. ein entsprechendes Merkmal) in mehreren Ausprägungen (Allelen) vorkommt. Mutationen vergrößern also den Polymorphismus. Bei SNPs betrifft der Polymorphismus nur ein einziges Nukleotid (=Einzelbaustein des Erbmoleküls DNA).
Neue SNPs sollten sich dann in der Population durchsetzen, d. h. alternative Sequenzen sollten verschwinden und damit die Vielfalt der SNPs verringert werden. Aber genau das wurde nicht gefunden, es konnten also im Drosophila-Genom* keine Bereiche identifiziert werden, in denen die erwarteten Effekte (geringerer Polymorphismus) auftraten. In dieser Langzeitstudie mit sich sexuell fortpflanzenden Organismen läuft Evolution gemessen an den Erwartungen in viel geringerem Umfang ab.
*Genom (= gesamtes Erbgut)
Die Autoren prüfen und diskutieren verschiedene Erklärungen für diesen Befund, ohne dass sie selbst eine davon beim derzeitigen Kenntnisstand als überzeugend einstufen. Die Laborbeobachtungen zeigen also, dass Selektion die genetische Variation in sich sexuell fortpflanzenden Populationen nicht wie erwartet reduziert. Bisher gilt als Lehrmeinung, dass Selektion, insbesondere wenn sie stark ist, im Laufe der Zeit zu deutlicher Verringerung des Gen-Polymorphismus, also der genetischen Vielfalt führt. Das konnten Burke et al. (2010) in ihrer Studie mit den Langzeit-Experimenten an Drosophila aber gerade nicht belegen. Das bedeutet, dass mit diesen experimentellen Resultaten der Einfluss von Selektion – bei der natürlichen Selektion handelt es sich um einen zentralen Evolutionsmechanismus – nicht bestätigt werden konnte.1
Da unter natürlichen Bedingungen die Selektionskriterien weniger stark und nicht über viele Generationen gleichbleibend ausgeprägt sind, kann man davon ausgehen, dass der ursprünglich erwartete Effekt unter Freilandbedingungen noch weniger auftreten wird.
Damit ist ein grundlegender, bisher angenommener Mechanismus für die Entstehung neuer Arten durch Selektionsprozesse in Frage gestellt. Weitere Forschung sollte dazu beitragen, die Abläufe besser zu verstehen.
Quelle
Burke MK, Dunham JP, Shahrestani P, Thornton KR, Rose MR & Long AD (2010) Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature doi:10.1038/nature09352; auf einer Internetseite sind Äußerungen von an der Untersuchung beteiligten Wissenschaftlern zusammengestellt: Scientists decode genomes of precocious fruit flies. ScienceDaily (2010.09.19) http://www.sciencedaily.com/releases/2010/09/100916162537.htm.
Anmerkung
1 Die Zusammenfassung des Beitrags in Nature wurde vom Herausgeber bezeichnenderweise mit dem Titel: „Experimental evolution reveals resistance to change“ veröffentlicht.
Autor dieserNews: Harald Binder
© 2011, http://www.genesisnet.info/schoepfung_evolution/n159.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
30.10.10 Konvergente Kooption von Pax-Genen
Oder: Was einmal taugt, taugt auch mehrmals
Eine der überraschenden Entdeckungen der letzten Jahre ist die vielfache Verwendung derselben Gene in verschiedenen Zusammenhängen. Evolutionstheoretisch wird postuliert, dass Gene häufig neu und sogar mehrfach „zweckentfremdet“ wurden; man spricht von „Kooption“ oder „Rekrutierung“. Immer häufiger zeigt sich darüber hinaus, dass ähnliche oder auch unähnliche Gene mehrfach unabhängig (konvergent) für denselben Zweck kooptiert wurden. Wie kann es sein, dass dies durch einen Evolutionsvorgang erfolgt, der nicht an irgendwelchen Zielen orientiert ist, aber dennoch zu einem sehr ähnlichen Ergebnis gelangt? Beispielsweise muss nach neuen Befunden die mehrfache Kooption von Pax-Genen angenommen werden. Diese Gene spielen eine herausragende Rolle bei der ontogenetischen* Entwicklung der Augen. Über eine neue Untersuchung darüber berichtet dieser Artikel und erläutert die erheblichen Probleme, die sich einem natürlichen Evolutionsprozess entgegenstellen.
*ontogenetisch (= betrifft die individuelle Entwicklung von der Befruchtung an)
Bei der Ausbildung von Augen spielt eine Reihe von Steuergenen eine wichtige Rolle. Das bekannteste darunter ist Pax-6. Dieses Gen kontrolliert die Augenentwicklung auf höchster Ebene bei den Bilateria („Zweiseitentiere“, dazu gehören z. B. die Gliederfüßer und die Wirbeltiere). Pax-6 ist z. B. beim Linsenauge der Säugetiere nötig, um eine nachfolgende Kaskade von etwa 2500 Genen in Gang zu setzen, die für die Formbildung des Auges wichtig sind (Halder et al. 1995); es wird daher auch als Masterkontrollgen bezeichnet. Pax-6 ist bei Arten, die zu ganz verschiedenen Tierstämmen gehören, so ähnlich, dass man es ohne Probleme zwischen sehr verschiedenen Tieren austauschen kann. So konnte die Arbeitsgruppe von Walter Gehring bereits 1995 zeigen, dass das Pax-6-Gen der Maus (welches hier eyeless heißt) auch in Fliegen seine Wirkung entfalten kann und die Bildung eines (Fliegen-)Auges auslöst (Halder et al. 1995).
Aufgrund dieser Ähnlichkeit wurde zunächst für alle Tiere mit Augen ein Vorfahr mit einem Urauge postuliert (Gehring 1996), dessen ontogenetische Bildung bereits mittels Pax-6 gesteuert wurde. Da das Komplexauge der Insekten und das Linsenauge der Wirbeltiere in ihrem Aufbau grundverschieden sind, muss das Urauge dieses postulierten gemeinsamen Vorfahren so einfach gedacht werden, dass es als Basis für die evolutionäre Entstehung beider Augentypen dienen kann. Bei einer völlig unabhängigen Entstehung von Komplexauge und Linsenauge müsste man alternativ annehmen, dass das Steuergen Pax6 erst sekundär und unabhängig aus einem anderen genetischen Funktionszusammenhang für die Augenentwicklung verwendet wurde, nachdem Komplex- und Linsenaugen evolutionär bereits entstanden waren. Dieser Prozess wird als extrem unwahrscheinlich betrachtet.
Kooption. Der hypothetische Vorgang der Inanspruchnahme vorhandener Gene für eine neue Aufgabe wird als „Kooption“ bezeichnet; man spricht auch von Gen-Rekrutierung oder auch von einer „Neuverschaltung“ von Genen. Die damit verknüpften Prozesse bzw. die einer solchen Neuordnung zugrunde liegenden Mechanismen sind aber weitgehend hypothetisch.
Da Kooption evolutionstheoretisch sehr unwahrscheinlich ist, muss man annehmen, dass Pax-6 schon sehr früh in der mutmaßlichen Evolution der Augen als Masterkontrollgen (s. o.) in Erscheinung trat, zu einem Zeitpunkt, als das Urauge noch sehr einfach und unspezialisiert gebaut war (s. o.).
Aufgrund der phylogenetischen Analysen (Ermittlung von Abstammungsverhältnissen durch Merkmalsvergleiche) stellte sich heraus, dass angesichts der genetischen Merkmalsverteilungen (hier Pax-Gene) zweifache oder sogar mehrfache konvergente Kooptionen gleicher Gene angenommen werden müssen. Das heißt: Die gleichen oder ähnlichen (Pax-)Gene wurden zweimal oder mehrfach unabhängig in unterschiedlichen stammesgeschichtlichen Linien (Hydrozoa, Cubozoa, Bilateria) in gleicher Weise für eine identische Funktion rekrutiert. Angesichts der Schwierigkeiten, die sich schon bei einer erfolgreichen Integration eines Gens in einen anderen Funktionszusammenhang stellt, wirft die Erklärung einer mehrfach unabhängigen gleichartigen Neu-Inanspruchnahme desselben Gens erst recht Fragen nach den Mechanismen auf. Mit der Zunahme der zu postulierenden Kooptionen (z. B. wie sie auch bei den Crystallinen für die Augenlinsen notwendig ist) wurde mehr und mehr von der Vermutung einer monophyletischen* Entstehung der Augen Abstand genommen (Gregory 2008, Piatigorsky 2008).
*monophyletisch (= von einem einzigen gemeinsamen Vorfahren abstammend)
Fünf Pax-Unterfamilien. Die Vertreter der Pax-Gengruppe werden in fünf Unterfamilien unterteilt. Pax-Gene besitzen in vielen Organismen zentrale Steuer- bzw. Schalterfunktionen in der Ontogenese verschiedenster Organe. Wie oben erwähnt, ist Pax-6 für die Augenentwicklung bei den Bilateria (Zweiseitentiere) entscheidend. Pax-6 kommt aber bei den Nesseltieren (Cnidaria) nicht vor. Interessanterweise haben vier der fünf Klassen der Nesseltiere, zu denen auch drei Klassen von Quallen gehören, Augen, z. T. sogar hochentwickelte Linsenaugen. Kürzlich veröffentlichten Suga et al. (2010) eine Untersuchung über das Vorkommen von Pax-Genen bei der Qualle Cladonema radiatum aus der Klasse der Hydrozoa. In diesem Tier wurden drei Pax-Gene, jeweils einer anderen Unterfamilie angehörig, Pax-A, Pax-B und Pax-E, gefunden. Suga et al. (2010) konnten zeigen, dass Pax-A bei der Bildung der Linsenaugen von Cladonema eine führende Rolle zukommt. Bei Tripedalia, die zu der Klasse Cubozoa (Würfelquallen) gehört, wurde in einer früheren Untersuchung Pax-B als verantwortlich für die Augenentwicklung nachgewiesen. Bei den Bilateria, den Hydrozoa und den Cubozoa werden also Pax-Gene aus verschiedenen Pax-Unterfamilien verwendet.
Suga et al. vermuten, dass die verschiedenen Pax-Unterfamilien durch Genduplikationen in einer sehr frühen Phase der Evolution aus einem urtümlichen Pax-Genvorläufer in einem augentragenden gemeinsamen Vorfahren entstanden sind, spätestens vor der Trennung der Bilateria und der Cnidaria (S. 14267). Es sei daher sehr wahrscheinlich, dass in den drei Linien der Hydrozoa, Cubozoa und Bilateria diese drei verschiedenen Pax-Gene zunächst unterschiedlich erhalten blieben (Pax-A und Pax-B fehlt bei den Bilateria, Pax-6 bei den Cnidariern) und dann unabhängig für die jetzige analoge Aufgabe in der strukturellen Augenentwicklung kooptiert wurden. (Andernfalls sollte man wohl bei allen Gruppen dasselbe Pax-Gen erwarten.) Die Autoren vermuten darüber hinaus, dass die kooptierten Pax-Gene im gemeinsamen Vorfahren der Cnidarier und Bilaterier bereits überschneidende Aufgaben bei der Augenentwicklung hatten. Damit sehen sie die monophyletische Entstehung aller Augentypen wieder als mögliche und gut begründete Hypothese im Rennen.
Bemerkenswert ist die Art und Weise, wie die Autoren die vermuteten Kooptionen beschreiben: Sie sprechen einerseits von einer Wahl unterschiedlicher Pax-Gene in den verschiedenen Tierklassen (S. 14267) und bezeichnen andererseits den hypothetischen Vorgang der Nutzung verschiedener Pax-Gene als „molecular-level opportunism“, den es häufig auch bei den Crystallinen (den Linsen-Proteinen) gegeben habe. Gemeint ist damit, dass in unterschiedlichen Linien zwar ähnliche, aber eben nicht identische Gene für einen bestimmten Zweck rekrutiert wurden. Die neuen Untersuchungen legen diesen Vorgang bei den Pax-Genen nahe. Es gibt aber auch Fälle, bei denen ganz unterschiedliche Gene rekrutiert werden, das ist bei den Linsencrystallinen vielfach der Fall (Piatigorski 2008).
Hinter diesen Beschreibungen stehen aber schwerwiegende Probleme für einen ungerichteten, natürlichen evolutionären Prozess. Schon eine einzige Übernahme eines Gens in ein neues, komplexes Netzwerk von Beziehungen erfordert vielfache Abstimmungen, insbesondere wenn es sich wie bei den Pax-Genen um Steuergene handelt, die eine prominente Position in den Regulationskaskaden einnehmen. Die Erklärungsprobleme potenzieren sich, wenn Kooptionen (wie im Falle von Pax-A, Pax-B und Pax-6) mehrfach unabhängig in ähnlicher Weise zum selben spezifischen Zweck der Strukturentwicklung des Auges erfolgt sein sollen.
Constraints. An dieser Stelle wird manchmal argumentiert, dass ähnliche Konstellationen bestimmte Kooptionsvorgänge begünstigen könnten; man spricht dann von „Kanalisierung“: Entwicklungszwänge, sog. constraints lenkten die weitere evolutive Entwicklung in eine bestimmte Richtung, wodurch es zu den Konvergenzen komme. Dies mag nachvollziehbar sein, wenn es um Feinabstimmungen und Spezialisierungen geht. Hier geht es aber um eine Neuverschaltung innerhalb eines bereits bestehenden komplexen genetischen Netzwerks. Der Hinweis auf constraints bietet keine Erklärung, da an diesem konkreten Beispiel der Augenentwicklung nicht gezeigt werden kann, worin diese bestehen sollen und wie sich diese auf genetischer oder morphologischer Ebene darstellen.
Die Annahme von constraints, die eine Kanalisierung und Parallelentwicklung in die Wege leiten, bleibt rein hypothetisch, solange nicht gezeigt wird, wie eine vorausgegangene Konstellation die nachfolgende Kooption begünstigt haben könnte. Wie soll ein Pax-Steuergen, das im Gesamtgefüge der Genwirkungen zunächst irgendeine andere Aufgabe unabhängig vom Sehen hatte, dafür prädestiniert gewesen sein, so dass es mit einer scheinbaren Notwendigkeit als Masterkontrollgen für die Bildung eines Auges kooptiert wurde? Pax-Gene haben verschiedene Funktionen – warum wurden sie mehrfach (d. h. mindestens zweimal oder dreimal) gerade für die Sehfunktion kooptiert? Warum Pax-Gene aus verschiedenen Familien? Da die Pax-Gene an prominenter Stelle wirken, braucht eine Kooption nach allem, was bekannt ist, wie erwähnt viele Abstimmungen. Wie kann dies dreimal unabhängig in unterschiedlichen evolutionären Stammeslinien in ähnlicher Weise mit ähnlichem Ergebnis ohne Planung so erfolgreich ablaufen?
Literatur
Gehring W (1996) The master control gene for morphogenesis and evolution of the eye. Genes Cells 1, 11-15.
Gregory R (2008) The evolution of complex organs. Evo. Edu. Outreach 1, 358-389.
Halder H, Callaerts P & Gehring WJ (1995) Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267, 1788-1792.
Piatigorski J (2008) A Genetic Perspective on Eye Evolution: Gene Sharing, Convergence and Parallelism. Evo. Edu. Outreach 1, 403-414.
Suga H, Tschoppa P, Graziussia DF, Stierwald M, Schmid V & Gehring WJ (2010) Flexibly deployed Pax genes in eye development at the early evolution of animals demonstrated by studies on a hydrozoan jellyfish. Proc. Natl. Acad. Sci. 107, 14263-14268.
Autor dieserNews: Reinhard Junker
© 2010, http://www.genesisnet.info/schoepfung_evolution/n156.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
20.10.10 Spezialisierter Sauerstofftransport: zweimal unabhängig „erfunden“
Aufgrund von phylogenetischen (= stammesgeschichtlichen) Analysen muss angenommen werden, dass der spezialisierte Sauerstofftransport zweimal unabhängig (= konvergent) entstanden sein muss: bei den Rundmäulern und den Kiefermündern. Dies ist bemerkenswert, weil mehrere ähnliche Vorgänge mit ähnlichem funktionalem Ergebnis angenommen werden müssen. Gene müssten unabhängig verdoppelt und für die neue Funktion rekrutiert worden sein; die durch sie codierten Hämoglobine weisen unabhängig ähnliche Spezialisierungen und biochemische Besonderheiten auf. Bekannte Evolutionsmechanismen scheinen für den erforderlichen hypothetischen Evolutionsprozess deutlich überfordert zu sein.
Der hypothetische Vorgang der Inanspruchnahme vorhandener Gene für eine neue Aufgabe wird als „Kooption“ bezeichnet, man spricht auch von Gen-Rekrutierung oder auch von einer Neuverschaltung von Genen. Solche Vorgänge sollen sich in der Evolution der Lebewesen häufig abgespielt haben. Die dafür erforderlichen Prozesse bzw. die einer solchen Neuordnung zugrunde liegenden Mechanismen sind aber weitgehend hypothetisch.
Immer mehr stellt sich nun aufgrund von phylogenetischen Analysen (Ermittlung von Abstammungsverhältnissen durch Merkmalsvergleiche) heraus, dass angesichts der genetischen Merkmalsverteilungen zweifache oder sogar mehrfache konvergente Kooptionen gleicher Gene angenommen werden müssen. Das heißt: Die gleichen oder ähnlichen Gene wurden zweimal oder mehrfach unabhängig in unterschiedlichen stammesgeschichtlichen Linien in gleicher Weise für eine identische Funktion rekrutiert. Angesichts der Schwierigkeiten, die sich schon bei einer erfolgreichen Integration eines Gens in einen anderen Funktionszusammenhang stellt, wirft die Erklärung einer mehrfach unabhängigen gleichartigen Neu-Inanspruchnahme desselben Gens erst recht Fragen nach den Mechanismen auf. Konvergente Kooptionen scheinen zudem keine seltenen Ausnahmen zu sein. Das in der Evolutionsforschung bekannte Konvergenzproblem (vgl. Ähnlichkeiten in der Morphologie und Anatomie, https://www.genesisnet.info/index.php?Artikel=41301&Sprache=de&l=1) begegnet einem auch hier. Wie kommt es, dass unabhängig voneinander (d. h. konvergent) dieselben bzw. ähnliche, aber auch unterschiedliche Gene für neue und zugleich identische Zwecke oder Funktionen in stammesgeschichtlich getrennten Linien genutzt werden?
Ein Beispiel einer konvergenten Kooption beschreiben Hoffmann et al. (2010). Es geht dabei um Hämoglobine, Moleküle, die für den Sauerstofftransport im Blut bei Wirbeltieren und einigen Wirbellosen sorgen. Die Evolution des Sauerstofftransports durch die Hämoglobine wird von den Autoren als „physiologische Schlüsselinnovation“ gewertet, ohne die größere Tiere nicht existieren könnten, da bloße passive Diffusion von Sauerstoff im Blutplasma nicht zur Versorgung der Gewebe ausreichen würde. Eine großangelegte phylogenetische Analyse der Globin-Gen-Superfamilie bei Wirbeltieren zeigte u. a., dass die Untergruppen der Hämoglobine in den Roten Blutkörperchen von Rundmäulern (Cyclostomata) und Kiefermündern (Gnathostomata, eine Überklasse der Wirbeltiere) nicht von orthologen* Genen kodiert werden, d. h. nicht durch die klassischen Artbildungsprozesse aus einem gemeinsamen Vorläufer entstanden sein können. Anders ausgedrückt: Aufgrund der den Globinen zugrundeliegenden genetischen Sequenzverteilungen können die beiden Hämoglobingruppen nicht von einem gemeinsamen Vorläufermolekül abgeleitet werden. Dennoch sind sie funktionell und teilweise in ihrem komplexen strukturellen Aufbau erstaunlich ähnlich.
*ortholog (= durch Artaufspaltung entstandene)
Dieser Tatbestand wirft die Frage auf, wie und in welchen Linien die aktuell funktionell ähnlichen, aber genetisch und biochemisch-strukturell deutlich unterschiedlichen Sauerstofftransportproteine von Rundmäulern und Kiefermündern phylogenetisch entstanden sind. Als Lösung des Problems wird von den Autoren zunächst vorgeschlagen, dass bei dem hypothetischen Vorläufer der Wirbeltiere verschiedene Vorläufervarianten der Globin-Gen-Superfamilie vorhanden waren, von denen die heute beobachtbaren verschiedenen und stark divergierenden Globin-Familien (z. B. die Hämoglobine) abzuleiten sind. Nach Trennung der Rundmäuler von den Kiefermündern erfuhren die verschiedenen genetischen Vorläufervarianten der Globingene eine unterschiedliche weitere Entwicklung. Einige Gene wurden mehrfach dupliziert (verdoppelt) und in neue Funktionszusammenhänge integriert andere verschwanden vollständig aus dem genetischen Repertoire (vgl. Abb. 1).
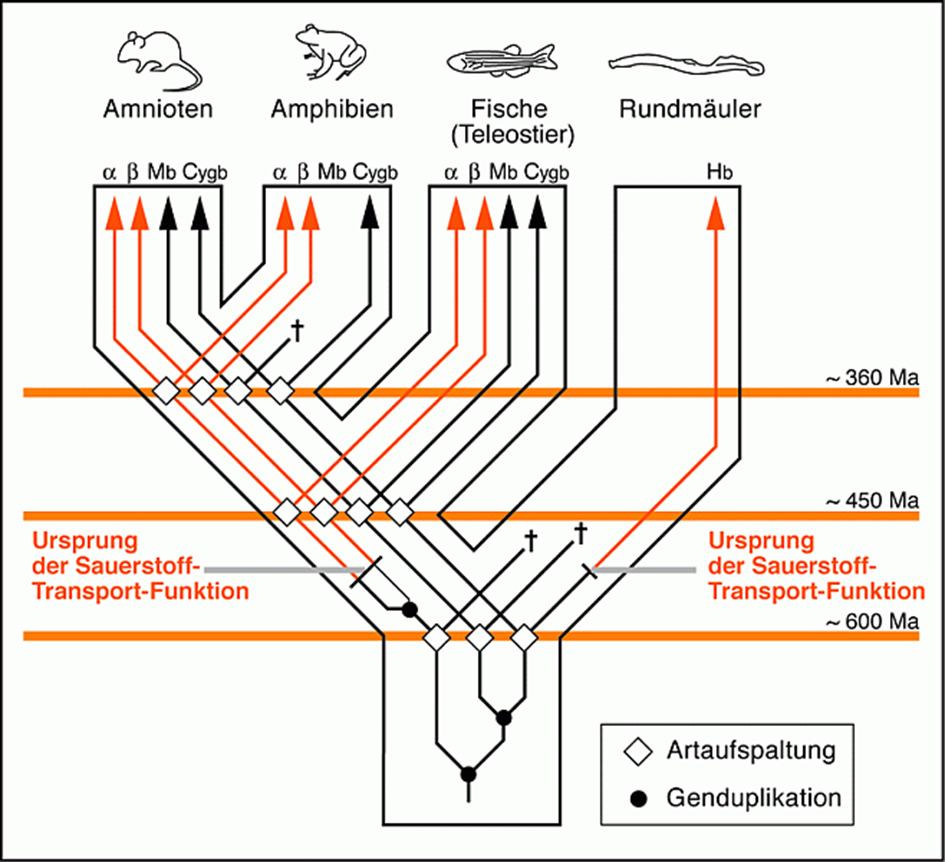
Abb. 1: Vermutete Duplikationen bei Globinen. Der Sauerstofftransport durch Hämoglobin soll zweimal unabhängig mit ähnlichen Mechanismen entstanden sein, bei den Rundmäulern und bei den Kiefermündern. Näheres im Text. (Nach Hoffmann et al. 2010)
Speziell für die Entwicklung der Hämoglobingruppen vermuten die Autoren einen Prozess, den sie „konvergente Kooption“ nennen. Für die erforderliche Funktion des Sauerstofftransports wurden bei den Rundmäulern und den Kiefermündern unterschiedliche „freie“ Kopien von speziellen Globin-Genen verwendet, die zuvor durch Genduplikation entstanden waren (die so entstandenen Gene werden als paralog bezeichnet). Die Integration der frei gewordenen Genduplikate in den neuen Funktionszusammenhang des Sauerstofftransports (Kooption) fand dann in den stammesgeschichtlich getrennten Linien ebenso wie die zuvor erforderliche Duplikation unabhängig voneinander statt und führte zu einem erstaunlich ähnlichen (konvergenten) Ergebnis. „Kooptive Konvergenz“ im Sinne der Autoren bedeutet also anders ausgedrückt, dass paraloge* Mitglieder derselben Globin-Genfamilie in den beiden Linien unabhängig voneinander dieselbe Spezialisierung1 der Funktion (Sauerstofftransport) und ähnliche biochemische Eigenschaften bei den durch sie codierten Hämoglobin-Proteine im Verlauf der Evolution hervorbrachten (S. 14274f.).
*paralog (= durch Duplikation entstanden)
Was wurde zweimal „erfunden“? Um das Ausmaß der evolutionstheoretisch anzunehmenden Konvergenz deutlich zu machen, muss die Funktionsweise des Hämoglobins (Hb) etwas erläutert werden: Bei den Kiefermündern ist Hb vierteilig, aus 2 a- und 2 b-Globin-Ketten aufgebaut. Hb kann vier Sauerstoffmoleküle aufnehmen, wobei die Aufnahmefähigkeit mit der Anzahl der bereits gebundenen Sauerstoffmoleküle zunimmt. Diese sogenannte positive Kooperativität der Sauerstoffbindung resultiert aus einer Veränderung der Quartärstruktur.
Das Hb der Rundmäuler besteht ebenfalls aus mehreren (zwei oder vier) Globin-Untereinheiten. Die Kooperativität der Sauerstoffbindung resultiert jedoch aus einer durch die Oxygenierung* verbundenen Trennung der Multimeren in gebundene Monomere. Die Hämoglobine sind in beiden Gruppen in Rote Blutkörperchen eingebunden und ermöglichen einen hocheffizienten Sauerstofftransport vom Aufnahmeort zu den Zielorten und tragen zum Rücktransport von CO2 bei. Die Effizienz beider Hämoglobine als Sauerstofftransportproteine wird durch eine Wechselwirkung der Untereinheiten erreicht (homotroper Effekt), die zur Kooperativität (s. o.) der Sauerstoffbindung führt. Die Sauerstoffabgabe im Gewebe erfolgt durch Bindung mit einem Liganden, die zu einer Konformationsänderung** führt (heterotroper Effekt). Sowohl im Hb der Rundmäuler als auch im Hb der Kiefermünder werden die Kooperativität und die Regulation durch Konformationsänderung aufgrund von Veränderungen der Quartärstruktur infolge der Oxygenierung ermöglicht (S. 14274f.).
*Oxygenierung (= Prozess der Sauerstoffbindung)
**Konformationsänderung (= Änderung der räumlichen Struktur)
Konvergent entstanden sind somit die Fähigkeit der Sauerstoffaufnahme und -abgabe sowohl durch homotrope als auch heterotrope kooperative Effekte. Die Kooperativität wird in beiden Fällen durch die Verbindung mehrerer Untereinheiten zu einer Quartärstruktur ermöglicht, wenn auch auf verschiedene Weisen. Diese Vorgänge müssten nach Hoffmann et al. in beiden Fällen von verschiedenen Globin-Monomeren ausgegangen sein, denen diese Kooperativität fehlte (14275). Abb. 1 oben verdeutlicht das mutmaßliche evolutionstheoretische Szenario.
Dass die beiden Hämoglobine nicht von einem gemeinsamen Vorläuferglobin mit Sauerstofffunktion stammen, schließen die Autoren aus zahlreichen Unterschieden in den strukturellen Details. Die Mechanismen der Sauerstoffaufnahme und -abgabe sind verschieden, beruhen aber in beiden Fällen auf Kooperativität mehrerer Untereinheiten. Die Autoren weisen darauf hin, dass die Hämoglobine beider untersuchten Gruppen zwar funktionell gleichartig, aber trotz der gleichen Benennung als Hämoglobine nicht homolog seien (S. 15277).
Jenseits der empirischen Wissenschaft. Die Autoren schließen ihren Artikel wie folgt: „Dieses Beispiel von konvergenter Evolution einer Proteinfunktion demonstriert eindrucksvoll die Fähigkeit der natürlichen Selektion, komplexe Design-Lösungen dadurch zusammenzuschustern, indem verschiedene Variationen desselben Protein-Gerüsts zusammengebastelt werden“ (S. 14278).
Angesichts der Tatsache, dass im gesamten Artikel von der Wirksamkeit der Selektion und den molekulargenetischen Mechanismen auf die mutmaßlichen Abläufe („konvergente Kooption“) nicht die Rede war, ist dieser Satz als Glaubenssatz zu werten und kein Ergebnis von empirischer Forschung. Er ist vielmehr Ausdruck von stillschweigenden Voraussetzungen: Funktionelle und/oder strukturelle Ähnlichkeiten (hier der Globine) sind abstammungsbedingt und der zentrale Mechanismus der Evolution ist die natürliche Auslese. Die von den Autoren vorgestellten Befunde sprechen diese Sprache aber nicht unbedingt, sie sprechen von einer unabhängigen „Erfindung“, natürlich ohne an einen Erfinder zu denken. Erstaunlich ist, dass man annehmen muss, dass der Selektionsdruck (hoher Sauerstoffbedarf bei größeren stoffwechselaktiven Tieren) dazu führt, dass die „Natur“ oder die „Evolution“ das gleiche funktionelle Ergebnis mit ähnlichen Abläufen der Sauerstoffaufnahme und -abgabe zweimal unabhängig voneinander hervorbringt. Die klassischen Mechanismen, wie sie im Rahmen der Erweiterten Synthetischen Evolutionstheorie diskutiert werden, können dieses Geschehen nicht plausibel machen. Neue Begriffe wie „konvergente Kooption“ erklären nichts, sondern verschleiern das eigentlich Unerwartete und Widersprüchliche der evolutionären Perspektive.
Nimmt man eine Schöpfungsperspektive ein (ebenfalls eine nicht allein aus den Daten ableitbare, aber mögliche Perspektive), ist durchaus verständlich, dass ähnlichen funktionellen Erfordernissen ähnliche Lösungen entsprechen. Diese sind im Detail verschieden, was aber auch wiederum im Detail den verschiedenen Anforderungen in den jeweiligen Organismen Rechnung tragen könnte. Unser Wissen ist bisher zu begrenzt, um dies beurteilen zu können.
Literatur
Hoffmann FG, Opazo JC & Storz JF (2010) Gene cooption and convergent evolution of oxygen transport hemoglobins in jawed and jawless vertebrates. Proc. Natl. Acad. Sci 107, 14274-14279.
Anmerkung
1 Es könnte sich auch um einen Neuerwerb der Funktion handeln. Die Autoren verweisen darauf, dass die verwendeten Globine vielleicht bereits irgendwie mit dem O2 Transport zu tun hatten. Das ist allerdings spekulativ.
Autor dieser News: Reinhard Junker (Unter Mitarbeit von Henrik Ullrich)
© 2010, http://www.genesisnet.info/schoepfung_evolution/n155.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
29.04.10 Der Ursprung des Insektenflügels
Eine japanische Forschergruppe berichtete kürzlich über entwicklungsgenetische Untersuchungen zur ontogenetischen* Bildung der Insektenflügel (Niwa et al. 2010). Aus ihren Ergebnissen schließen sie, dass einige Bauplanänderungen, die zur Ausbildung des häutigen Insektenflügels erforderlich waren, durch relativ wenige Änderungsschritte möglich gewesen sein könnten. Die von den Autoren vorgestellten Befunde aus der Entwicklungsbiologie sind jedoch keineswegs ausreichend für die Erklärung des evolutionären Werdens von Insektenflügeln, sondern nur Randbedingungen, die beim Versuch einer evolutionstheoretischen Erklärung berücksichtigt werden müssen.
*ontogenetisch (= Entwicklung von der Eizelle bis zum ausgewachsenen Organismus)
Einleitung. Wie entstanden ganz neue Bauelemente der Lebewesen wie z. B. Flügel? Woher kommen evolutive Neuheiten? In den letzten Jahren wurden zahlreiche erstaunliche Entdeckungen in der Entwicklungsbiologie gemacht, von denen man sich Antworten auf diese Frage erhofft. In der Entwicklungsbiologie geht es um Vorgänge, die zum Wachstum und zur Differenzierung des individuellen Organismus führen, ihr Forschungsgegenstand ist also die Entwicklung von der Befruchtung bis zum ausgewachsenen Organismus (Ontogenese). Im Zeitalter der Molekulargenetik geht es dabei besonders um die Rolle der (Regulations-)Gene im Zellwachstum und in der Spezialisierung und Differenzierung der Zellen, in der Ausbildung der Organanlagen und ihrer Ausformung. In der Evolutionären Entwicklungsbiologie (kurz: Evo-Devo) versucht man, aus der Erforschung der Regulationsprozesse in der Ontogenese Hinweise für die hypothetische Phylogenese (Stammesgeschichte, Evolution) zu gewinnen. Zur Evolutionären Entwicklungsbiologie siehe den Artikel Evo-Devo, https://www.genesisnet.info/index.php?Artikel=41266&Sprache=de&l=2.
Weshalb wird in die evolutionäre Entwicklungsbiologie die Hoffnung gesetzt, dass mit ihrer Hilfe Fragen nach dem Ursprung evolutiver Neuheiten geklärt werden könnten? Es sind unter anderem die Entdeckung von Regulationsgenen, von Mehrfachnutzung von Genen oder ganzen Entwicklungsmodulen sowie des teilweise modularen Aufbaus der Netzwerke zwischen den Erbinformationen und zahlreichen zellulären Prozessen und Entwicklungsvorgängen. Manche Evolutionsbiologen sehen darin das Potential, dass durch Neuverschaltungen von Genen, insbesondere Regulationsgenen, oder ganzer Entwicklungsmodule neue Organe relativ schnell entstehen können. Diese Möglichkeit wird zum Beispiel bei der Entstehung des Schildkrötenpanzers oder des Fledermausflügels diskutiert; und jüngst am Beispiel der Entstehung des Insektenflügels, um das es im Folgenden gehen soll.
Der Ursprung des Insektenflügels. Eine japanische Forschergruppe um Shigeo Hayashi berichtete kürzlich über entwicklungsgenetische Untersuchungen zur ontogenetischen Bildung der Insektenflügel (Niwa et al. 2010). Aus ihren Ergebnissen schließen sie, dass einige Bauplanänderungen, die zur Ausbildung des häutigen Insektenflügels erforderlich waren, durch relativ wenige Änderungsschritte möglich gewesen sein könnten. Es handelt sich dabei um ein typisches Beispiel dafür, wie man versucht, die Ergebnisse der Entwicklungsbiologie für die Lösung evolutionstheoretischer Fragestellungen zu nutzen. Es soll in diesem Artikel jedoch gezeigt werden, dass die Erklärungskraft bezüglich evolutionärer Hypothesen sehr begrenzt ist: Die von den Autoren vorgestellten Befunde aus der Entwicklungsbiologie sind keineswegs ausreichend für die Erklärung des evolutionären Werdens von Insektenflügeln, sondern sind nur Randbedingungen, die beim Versuch einer evolutionstheoretischen Erklärung berücksichtigt werden müssen.
Die Entstehung des Insektenflügels konnte durch Fossilfunde nicht erhellt werden, „da die ältesten bekannten Fossilien geflügelter Insekten (Pterygota) bereits über einen hochentwickelten, voll ausgebildeten Flugapparat mit großen Flügeln und komplexer Flügeladerung verfügen“ (Hörnschemeyer 2009). Fossilien, die Übergänge zeigen, sind nicht bekannt. Hypothesen zur evolutiven Entstehung des Insektenflügels beruhen daher auf vergleichenden Studien heute lebender oder fossiler Formen. Zur Entstehung des Insektenflügels werden zwei Hypothesen diskutiert (vgl. Hörnschemeyer 2009, Niwa et al. 2010):
- Epicoxal-Theorie: Die Flügel sind aus dem beweglichen äußeren Anhang des obersten Beingliedes (der Epicoxa) der ursprünglichen Gliederfüßer-Beine hervorgegangen. Der Vorteil dieser Theorie besteht darin, dass bereits Muskulatur vorhanden ist. Problematisch sind dagegen u. a. die Entstehung von flachen Tragflächen und der rückenseitigen Lage der Flügel.
- Paranotaltheorie. Die Insektenflügel sind als seitliche Abfaltungen im Brustsegment bzw. im Grenzbereich Rücken-Seite entstanden. Diese Theorie hat den Vorteil, dass die Flächigkeit leichter verständlich ist; ein Problem von vielen ist aber die Entstehung der Flugmuskulatur und der Gelenke, die nach dieser Theorie de novo erfolgt sein müssten.
Die Untersuchungen von Niwa et al. und ihr Modell. Niwa et al. (2010) untersuchten drei für die Flügelausformung wichtige Regulatorgene (wingless, wg, vestigial, vg und apterous, ap) bei Vertretern zweier Insektengruppen, die als evolutionär ursprünglich eingestuft werden, nämlich bei den Eintagsfliegen (Ephemeroptera) und bei ungeflügelten Felsenspringern (Archaeognatha). Diese Insekten besitzen rückenseitig Körperanhänge, und zwar Tracheenkiemen (Nymphen der Eintagsfliege) bzw. sogenannte Styli (Felsenspringer), das sind kurze, stabförmige Auswüchse an der Körperseite von deren Nymphen. Die Untersuchungen der Wissenschaftler ergaben, dass zwei Regulationsmodule, die mit wg und vg verknüpft sind und die für die Ausbildung des Flügels benötigt werden, auch in den flügellosen Insekten vorkommen: Das eine ist im Bereich der seitlichen Körperregion aktiv und induziert stabförmige Beinanhänge, das andere im Bereich der Grenze zwischen Rücken und Seite und ist mit dem flachen Auswuchs in deren Grenzbereich verbunden.
Niwa et al. (2010, 174) schlagen folgendes Modell der Flügelentstehung vor: Zwei Regulationsgene, die in der ontogenetischen Entwicklung der Flügel beteiligt sind (nämlich wg und vg), hatten in den mutmaßlichen Vorfahren der geflügelten Insekten eine andere Funktion, nämlich in der Induktion des oberen, stäbchenförmigen Asts der verzweigten Gliedmaßen bzw. in der Induktion von flachen Auswüchsen an der Grenze von Rückenschild und seitlicher Körperregion (Fig. 5 in Niwa et al.). Durch Kombination dieser der beiden Entwicklungsmodule und die Verschiebung ihres Wirkungsbereichs in der seitlichen Körperregion sei die rasche Entstehung des Flügels begünstigt worden. Dadurch sei außerdem die flache Form der seitlichen Körperauswüchse mit der Beweglichkeit der Gliedmaßen kombiniert worden.
Dieses Modell ist zugleich eine Kombination der Epicoxal- und der Paranotaltheorie (s. o.). Typisch für „Evo-Devo“ ist die Idee, dass sich durch wenig geänderte Verschaltungen von Genen und ihrer Wirkungsbereiche neue Organe bilden könnten (hier der Flügel von Insekten) und darüber hinaus, dass dies in relativ kurzen Zeitabschnitten vonstatten gehen könnte (womit sich die Suche nach Übergangsformen erübrigen würde). So spricht Hemminger (2010) von einer „einfachen Integration“ der beiden vorher getrennten Steuer-Module und kommentiert: „Das vorliegende Modell zeigt, dass sich selbst große Bauplanänderungen, … auf der Ebene der Entwicklungssteuerung durch relativ wenige Veränderungsschritte erklären lassen“ (Hervorhebung nicht im Original).
Was haben Niwa et al. gezeigt? Bei der hier anstehenden Frage, wie ein Organ entsteht, geht es um Mechanismen und Abläufe. Die Untersuchungen von Niwa et al. haben aufgedeckt, dass für die Induktion des Stylus bei Felsenspringern, der Tracheenkiemen bei Eintagsfliegenlarven und bei den Flügeln der Taufliege teilweise gleiche Entwicklungsmodule genutzt werden – zweifellos eine sehr interessante Entdeckung durch wegweisende Experimente. Wie diese gleichen Module für die verschiedenen Aufgaben – hier die Induktion verschiedener Körperanhänge – rekrutiert und verschaltet wurden, wird durch diese Gemeinsamkeiten aber überhaupt nicht geklärt. Die Befunde von Niwa et al. liefern also eine wichtige Basis für das von ihnen vorgeschlagene Entstehungsmodell. Das Verständnis der ontogenetischen Zusammenhänge ist aber als solches nicht schon die Erklärung des hypothetisch vorgeschlagenen Evolutionsvorgangs. Die Autoren haben interessante Bausteine geliefert, die berücksichtigt werden müssen, wenn man konkrete Hypothesen über evolutive Mechanismen des Umbaus hin zum Insektenflügel aufstellt. Sie äußern sich dabei selbst vorsichtig und unterscheiden zwischen den gewonnenen Daten und den Spekulationen über mögliche Evolutionswege.
Eine Erklärung müsste zeigen, wie durch ungerichtete Mutationen die postulierten Veränderungen eintreten, es bräuchte experimentelle Belege dafür, dass solche Verschaltungen von Modulen (auch schrittweise) möglich und funktional und selektionspositiv sind (andernfalls könnten sie nicht durchsetzen).1 Weiter müsste geklärt werden, welche Nebenwirkungen die vermuteten Neuverschaltungen hatten und ob sie tolerabel sind. Dieselben Gene (insbesondere die Masterkontrollgene) üben örtlich und zeitlich koordiniert zahlreiche Funktionen im sich entwickelnden Organismus aus und sind in einem komplizierten Netzwerk miteinander verbunden. Änderungen betreffen also viele Aspekte der Organisation der Lebewesen und ihrer Ontogenese.
Kurz: die postulierte Neuverknüpfung von Entwicklungsmodulen benötigt vielfache parallele Abstimmungen, damit sie selektionspositiv bzw. mindestens neutral sein kann. Mit „einfachen“ Änderungen auf der Ebene der Entwicklungssteuerung ist es deshalb bei weitem nicht getan. Die Integration eines neuen bzw. die Abwandlung eines vorhandenen Schalters in einem örtlich und zeitlich fein abgestimmten Netzwerk von Entwicklungsmodulen erzeugt nicht per se – wie übrigens auch in der Technik nicht – eine neue Funktionalität oder ein neues Bauprinzip des betroffenen Teilsystems.
Selbst wenn die mit der Verflechtung der Gene und Entwicklungsmodule verbundenen Probleme der Steuerung gelöst werden könnten, hätte man mit der Rekrutierung und Verschaltung zweier Regulationsgene und der Verschiebung ihres Wirkungsbereichs nicht einmal ansatzweise die Entstehung des Flügels erklärt. Denn der Flügel muss eine bestimmte Form besitzen, die Flügelmuskulatur muss entsprechend angepasst sein, die Verbindung des Flügels mit der Flugmuskulatur muss funktional sein, Gelenke müssen ausgebildet sein, das Insekt benötigt ein entsprechendes Verhalten und alle diese Vorgänge benötigen neue Steuerungsvorgänge in der Ontogenese. Ein Zitat zum Aufbau des Flügelgelenks kann die Problematik beispielhaft verdeutlichen: „Es handelt sich dabei um ein hoch komplexes System aus mehreren Skleriten, die in ihrem mechanischen Zusammenspiel dafür sorgen, dass die Bewegungen der Muskulatur auf den Flügel übertragen werden. Dazu gehören neben dem Auf- und Abschlag auch Steuerbewegungen und das Zusammenlegen oder Falten der Flügel“ (Hörnschemeyer 2009). Neben den bereits genannten organismusinternen Rahmenbedingungen eines Systemumbaus („constraints“) bleibt im theoretischen System der Synthetischen Evolutionstheorie die Frage offen, welche Selektionsdrücke den postulierten Umbau steuerten. Wurde in Richtung auf die Flugfähigkeit hin selektiert, wären die o. g. Eigenschaften des Flügels relativ kurzfristig und gleichzeitig erforderlich gewesen, um selektionspositiv zu sein. Wurde nicht in Richtung Flugfähigkeit selektiert, bleibt zu klären, welche selektionspositive Primärfunktionen die „Vor“-flügel besaßen, bevor diese zum Fliegen genutzt wurden und wann, warum und wie der Funktionswechsel erfolgte.
Bei allen diesen Überlegungen muss man sich vor Augen halten, dass Flügel von Insekten immer mehr als relativ simple Ausstülpungen sind, auch bei Insekten in Bernstein und bei den als „ursprünglich“ geltenden Eintagsfliegen. Dabei ist nichts „primitiv“! Besonders erstaunlich ist z. B. die Libellenflügel-Konstruktion. Hier werden die Flügel durch eine äußerst komplizierte Muskulatur unabhängig voneinander bewegt, während bei Wespen, Bienen, Fliegen der Auf- und Abschlag der Flügel synchron durch sich zusammenziehende Thoraxmuskulatur erfolgt. Jede Flügel-Muskulatur-Gelenk-Kombination ist für sich genommen ein technisch fein abgestimmtes Organ. Bei Bienen haken etwa die Hamuli (Häkchen am Vorderrand des Hinterflügels) von unten (!) in die entsprechend ausgebildete Randleiste des Vorderflügels ein, so dass beim Abschlag mit den größeren Kraftwirkungen der Hinterflügel vom Vorderflügel quasi flächig mit nach unten gedrückt wird. Beim Aufschlag wird der Hinterflügel mit weniger Kraft nach oben gezogen.
Die Konstruktion ist so ausgefeilt, dass wegen der im äußeren Bereich der Flügel auftretenden größeren Kräfte zweckmäßigerweise auch eine drastische Vergrößerung der Auflagefläche des Vorderflügels auf den Hinterflügel vorzufinden ist. Dies wird durch eine Verbreiterung des Vorderflügels im Außenbereich von dessen Hinterkante erreicht. So wird beim Abschlag verhindert, dass sich die Hamuli wie bei einem Reißverschluss vom Vorderflügel lösen (Abb. Winfried). Dazu kommt beim Bienenflügel die speziell gefertigte Muskulatur, die – gesteuert durch das Gehirn – dafür sorgt, dass sich der Hinterflügel nach dem Landen aus der Leiste des Vorderflügels löst und als Ganzes unter den Vorderflügel geschoben werden kann. (Zu den Insektenflügeln bzw. Flugsystemen siehe auch http://members.liwest.at/rammerstorfer/IflightIdesignPDF1.pdf.)
Ein einfacher und schneller Erwerb der Flugfähigkeit ist durch die Befunde von Niwa et al. also nicht einmal ansatzweise demonstriert. Die populäre Darstellung „But it turns out that just two genes, may explain insect wings“ von Dan Vergano (2010) in USA Today ist durch die Daten nicht im Entferntesten gedeckt. Die Autoren des Originalartikels sind im Gegensatz zur Darstellung bei Hemminger (2010) so vorsichtig, dass sie nur davon sprechen, dass die hypothetische Kombination der beteiligten Regulationsgene eine schnelle Entstehung des Flügels „erleichtert“ habe (S. 174). Macht man sich klar, was man für einen funktionsfähigen Flügel im Einzelnen benötigt, wird deutlich, dass diese „Erleichterung“ gegenüber den noch zusätzlich erforderlichen Änderungen und Innovationen auf dem Weg zum Insektenflügel nur als eine minimale Rahmenbedingung und nicht als Lösung des evolutionstheoretischen Problems betrachtet werden kann.
Dank: Wertvolle Hinweise erhielt ich von Winfried Borlinghaus, Jean-Luc Murk und Henrik Ullrich.
Anmerkung
1 Dabei muss natürlich nicht gleich die Flugfunktion erreicht werden, aber hypothetische Zwischenstufen auf dem Weg zur Flugfähigkeit müssen irgendeine Funktion ausüben.
Quellen
Hemminger H (2010) Die rasche Entstehung des Insektenflügels. http://ag-evolutionsbiologie.de/app/download/3323624202/evolution-der-insektenfluegel.html.
Hörnschemeyer T (2009) Evolution des Flugapparates der Insekten. http://www.uni-goettingen.de/de/text/87297.html.
Niwa N, Akimoto-Kato A, Niimi T, Tojo K, Machida R & Hayashi S (2010) Evolutionary origin of the insect wing via integration of two developmental modules. Evol. Dev. 12, 168-176.
Vergano D (2010) Insect wing evolution revealed in recycled genes. http://www.usatoday.com/tech/science/columnist/vergano/2010-03-26- wings29_ST_COLUMN_N.htm.
Autor dieserNews: Reinhard Junker
© 2010, http://www.genesisnet.info/schoepfung_evolution/n148.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
27.03.10 Muskelmasse: 2785 Gene für ihre Funktion
Wer kennt sie nicht, die Drosophila melanogaster, als Taufliege, Obstfliege oder Essigfliege bekannt. Man trifft sie nicht nur in jedem Haushalt, sondern das wenige Millimeter große Tier ist auch seit etwa hundert Jahren in den Labors der Biologen zuhause; mit ihm wurden unzählige Experimente durchgeführt. Eine interessante Studie zu den genetischen Grundlagen der Muskeln und ihrer Funktion veröffentlichte kürzlich eine Arbeitsgruppe um Frank Schnorrer vom Max-Planck-Institut für Biochemie in Martinsried.
Mit Hilfe einer speziellen Methode, der sogenannten RNAi-Technik (RNA-Interferenz; vgl. http://en.wikipedia.org/wiki/RNAi), hatten die Forscher systematisch alle 12.000 Gene der Taufliege analysiert. Die Gene wurden mit dieser Technik nach und nach einzeln ausgeschaltet und die Folgen für das Flugverhalten untersucht (Schnorrer et al. 2010). Es zeigte sich, dass 2785 Gene für die Muskelentwicklung und Muskelfunktion benötigt werden. Viele Gene konnten bestimmten Funktionen im Aufbau der Muskeln, der Muskelfibrillen oder der Sarkomere* zugeordnet werden. Die Forscher untersuchten zusätzlich zur RNAi-Technik auch Genmutationen, die zu Muskeldefekten führen.
*Sarkomere (= kleinste funktionelle Einheit der Muskelfibrille)
Da viele der bei Drosophila nachgewiesenen Gene auch beim Menschen vorkommen und dort wahrscheinlich ebenfalls für eine normale Muskelfunktion benötigt werden, könnte die Studie auch Bedeutung für das Verständnis der Genfunktionen der Muskulatur von Wirbeltieren haben und Einsichten für die Behandlung von Muskelerkrankungen ermöglichen.
„Ein Teil der entdeckten Gene wird in allen Muskeln gebraucht, ein anderer Teil nur in den sehr schnellen, sehr kraftvollen Flugmuskeln“, wird Schnorrer bei Spiegel online zitiert. Die Flugmuskeln der Insekten gehören zu den kräftigsten Muskeln im Tierreich: „Sie können bis zu 100 Watt pro Kilogramm Muskelmasse erzeugen und das über einen langen Zeitraum. Davon können Bodybuilder oder Tour-de-France-Fahrer nur träumen.“ Diese schaffen dauerhaft etwa 30 Watt pro Kilogramm Muskelmasse (http://www.spiegel.de/wissenschaft/natur/0,1518,682813,00.html).
Die Untersuchungen über die Muskelgene bei der Taufliege sind aber auch von grundsätzlicher Bedeutung, wenn es darum geht, evolutionäre Hypothesen zu entwickeln. Angesichts des auch hier wieder bestätigten Befundes, dass dieselben Gene bei sehr verschiedenen Tiergruppen Verwendung finden (hier bei Gliederfüßern und Wirbeltieren), wurde auch hier vorgeschlagen, dass vorhandene Gene in neue Zusammenhänge eingeflickt worden sind und dass auf diese Weise durch „Neuprogrammierung“ auch neue Konstruktionen entstehen könnten.1 Angesichts der großen Anzahl der für die (Flug-)Muskulatur benötigten Gene erweisen sich solche einfachen Vorstellungen als vollkommen unrealistisch. Denn ein (hypothetisches) Neuverschalten vorhandener Gene oder Entwicklungsmodule erfordert vielseitige Abstimmungen innerhalb der jeweiligen Vorkonstruktion. Ohne deren Kenntnis können Aussagen, die über sehr allgemeine Hypothesen zur Evolution hinausgehen, gar nicht gemacht werden.
Der Befund, dass über 20 % der etwa 12.000 Drosophila-Gene alleine für die Funktionen der Muskulatur benötigt werden, kann auch als Hinweis darauf gewertet werden, dass die Gene mehrfach in verschiedenen Zusammenhängen genutzt werden (man spricht von pleiotropen Genen, vgl. Piatigorsky 2007, xiii: „My original idea was to provide an extensive list of multifunctional proteins. I soon realized the futility of that approach: I would have to include most, if not all, proteins!“). Die damit einhergehende Vernetzung muss in evolutionären Hypothesen ebenfalls berücksichtigt werden und lässt die Vorstellung, wenige Mutationen könnten weitreichende konstruktive Änderungen mit positiven Eigenschaften ermöglichen, noch unglaubwürdiger erscheinen.
Anmerkung
1 Dies geht so weit, dass behauptet wird, sehr wenige Mutationen in Regulationsgenen wären z. B. die Entstehung des Fledermausflügels verantwortlich oder die Verschaltung von Modulen würde die Entstehung des Insektenflügels ermöglichen (Niwa et al. 2010).
Literatur
Niwa N et al. (2010) Evolutionary origin of the insect wing via integration of two developmental modules. Evolution & Development 12, 168-176.
Piatigorsky J (2007) Gene sharing and evolution. The diversity of protein functions. Cambridge, Mass. and London.
Schnorrer F, Schönbauer C, Langer CCH, Dietzl G, Novatchkova M, Schernhuber K, Fellner, Anna Azaryan M, Radolf M, Stark A, Keleman K & Dickson BJ (2010) Systematic genetic analysis of muscle morphogenesis and function in Drosophila. Nature 464, 287-291. Abstract unter http://www.nature.com/nature/journal/v464/n7286/abs/nature08799.html
Autor dieserNews: Reinhard Junker
© 2010, http://www.genesisnet.info/schoepfung_evolution/n145.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
11.12.09 Evolution bei Bakterien – neue Befunde
Neue Untersuchungen aus einem Langzeit-Evolutionsexperiment mit Escherichia coli zeigen, dass die Genome (= komplettes Erbgut) im Verlauf von 40.000 Generationen verkleinert werden, die Mutationen nicht einfach mit der Zahl der Generationen korrelieren und der Zusammenhang zwischen Änderungen im Genom und der Anpassung des Organismus komplex zu sein scheint. Die über 650 genetischen Veränderungen im Genom von E. coli in 40.000 Generationen verändern den Organismus (Phänotyp) nur wenig. Die Komplexität der Zusammenhänge erlaubt derzeit aber kein Urteil über prinzipielle Grenzen.
In der Nature-Ausgabe zum 200. Geburtstag von Charles Darwin vom 12. 2. 2009 veröffentlichten Buckling et al. (2009) eine zusammenfassende Arbeit über experimentelle Evolution mit Mikroorganismen. Mit diesen Kleinstlebewesen, die kurze Generationszeiten aufweisen (typischerweise mehrere Generationen/Tag), können spezifische Fragestellungen zu evolutionären Mechanismen im Labor unter definierten Bedingungen empirisch untersucht werden. Obwohl bereits zu Darwins Lebzeiten solche Experimente durchgeführt wurden, führten erst die Langzeit-Evolutionsexperimente (LZEE) mit Escherichia coli von Lenski und seinen Mitarbeitern zu größerer Aufmerksamkeit und Popularität solcher Untersuchungen.
Buckling et al. äußern die Zuversicht, dass Genome (= Ergbut) von Mikroorganismen nicht nur zu Beginn und am Ende eines Selektionsprozesses analysiert werden können, sondern dass auch Genome von Individuen aus den Zwischenschritten zugänglich werden.
Nun haben Lenski und Mitarbeiter (Barricke et al. 2009) eine Arbeit vorgelegt, in der sie mit modernen Methoden der Gensequenzierung komplette Genome aus verschiedenen Proben aus ihrem LZEE mit inzwischen über 40.000 Generationen von E. coli analysieren. In einem Begleitkommentar verweist Rainey (2009) auf die Bedeutung, die der Paläontologe Simpson 1944 in seinem einflussreichen Buch „Tempo and Mode in Evolution“ der Unterscheidung von Geschwindigkeit der evolutionären Änderungen und deren Mechanismen beimisst.
Barricke et al. analysieren die Geschwindigkeit und die Art und Weise der Evolution von Genomen aus LZEE nach 2000, 5000, 10.000, 15.000, 20.000 und 40.000 Generationen von E. coli. Die bisher verfügbaren Daten zeigen eine fast lineare Verteilung der Mutationen im Verlauf der ersten 20.000 Generationen. Nach ca. 26.500 Generationen steigt die Mutationshäufigkeit bis zur 40.000sten Generation deutlich stärker an. Dagegen nimmt die Steigerung der Fitness nach einem zunächst steilen Anstieg mit zunehmender Dauer ab, ein Hinweis darauf, dass die vorteilhaften genetischen Veränderungen (Mutationen) im Lauf der Zeit weniger werden. In 20 000 Generationen haben sich im Genom von E. coli 45 Mutationen manifestiert, darunter 29 einzelne Nukleotid-Austausche (SNPs) und 16 Deletionen (= Verluste), Insertionen (= Einschübe) und andere Polymorphismen. Die Autoren diskutieren diesen überraschenden Befund der unterschiedlichen Tendenz zwischen Mutation und Fitness vor dem Hintergrund verschiedener Evolutionsmodelle.
Das Genom der 40 000sten Generation weist im Vergleich zum Ausgangsgenom 627 SNPs und 26 weitere Polymorphismen auf. Eine Analyse zusätzlicher Proben bringt die Autoren zur Vermutung, dass der Einschub eines Nukleotids (Insertion) den Leserahmen für das mutT Gen verschiebt, was für die Erhöhung der Mutationsrate ursächlich ist. Dieses Ereignis – so legen es die Analysedaten nahe – hat sich irgendwann nach 25.000 Generationen abgespielt, bei Generation 26.500 ist die Mutation etabliert. Interessanterweise ist das Genom von E. coli nach 40.000 Generation verglichen mit dem Ausgangsgenom um 1,2 % reduziert und weist nur noch 4,57 x 106 bp (Basenpaare) auf.
Abschließend verweisen die Autoren auf das Potential der Analyse kompletter Genome im Zusammenhang mit Untersuchungen zur experimentellen Evolution. Sie weisen aber auch darauf hin, dass die komplexen Zusammenhänge zwischen Mutationsraten und Anpassung selbst in diesem vergleichsweise einfachen System keine allgemeine Erklärung von Änderungen im Genom unter natürlichen Bedingungen zulässt, ohne spezifische Erkenntnisse über die molekularen und Populationsgenetischen Prozesse einzubeziehen. Die überraschenden Ergebnisse dieser Untersuchung zeigen selbst in diesem vergleichsweise wenig komplexen System Kopplungen verschiedenster Einflüsse, die für eine Erklärung berücksichtigt werden müssen. Einfache Modelle haben sich hier als wenig hilfreich erwiesen und man darf auf weitere Erfahrungen aus dieser und anderen Experimentalreihen gespannt sein.
Die von Lenski und seinen Mitarbeitern hier veröffentlichen Befunde zeigen, dass der Zusammenhang zwischen Genom und Anpassung des Organismus an seine Umwelt von verschiedenen Faktoren abhängig sind, über die wir selbst in vergleichsweise einfachen Systemen bisher nur unzureichende Kenntnisse haben.
Literatur
Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE & Kim JF (2009) Genome evolution and adaption in a long-term experiment with Escherichia coli. Nature 461, 1243-1247.
Binder H (2008) Langzeit-Evolutionsexperiment mit Escherichia coli. Empirischer Befund für neue Funktion durch Mutation? Stud. Int. J. 15, 96-98.
Buckling A, Maclean RC, Brockhurst MA & Colegrave N (2009) The Beagle in a bottle. Nature 457, 824-829.
Rainey PB (2009) Arhythmia of tempo and mode. Nature 461, 1219-1221.
Autor dieserNews: Harald Binder
© 2009, http://www.genesisnet.info/schoepfung_evolution/n139.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
26.03.09 Von wegen Schrott – immer wieder neue Befunde aus dem Genom
Projekte zur Sequenzierung von Genomen haben gezeigt, dass ein erheblicher Anteil des Erbmoleküls DNA nicht in Proteine (= Eiweißverbindungen) übersetzt wird, in diesem Sinne also nicht codiert. Beim menschlichen Genom (= Erbgut) umfasst nicht-codierende DNA mehr als 95 % des Genoms. Die nicht-codierende DNA wurde von verschiedenen Autoren als „junk DNA“ bezeichnet, d. h. als Abfall der Evolution; eine Sichtweise, die sich zunehmend als voreilig und nicht zutreffend herausstellt.
In Genomprojekten wurden auch Methoden entwickelt, die es ermöglichen diejenigen DNA-Abschnitte zu bestimmen, die in RNA umgeschrieben (transkribiert) werden (Transkriptom). Dadurch rückten DNA-Abschnitte ins Blickfeld, die man als nicht-codierend bezeichnet, weil sie wie eingangs bereits erwähnt nicht durch Transkription und Translation in Peptidpolymere (= aus Aminosäuren gebildete kettenförmige Moleküle) übersetzt werden.
So wurden aus Bereichen nicht codierender DNA z. B. verschiedene Formen von microRNA (miRNA) beschrieben – kurze RNA Stränge (19-23 Nukleotide (= Einzelbausteine der DNA)) –, die durch Bindung an die mRNA die Genexpression (= Ablesen der Gen-Information) regulieren.
Als 1999 die komplette DNA-Sequenz vom Chromoson 22 – und ein Jahr später die erste Version des gesamten menschlichen Genoms – veröffentlicht worden war, untersuchten Rinn et al. (2003) diese Daten nach nicht-codierenden Abschnitten, die dennoch in RNA umgeschrieben (transkribiert) wurden und damit irgendeine Funktion signalisieren. Rinn und seine Kollegen konzentrieren sich seither auf große, eingestreute, nicht-codierende RNA-Abschnitte (large intervening non-coding RNAs; lincRNA). Die Befunde und deren Interpretation als RNA mit Funktion wurden in Fachkreisen durchaus kontrovers diskutiert. Kritiker vermuteten z. B. durch die Analysentechnik bedingte Fehler.
Der Autor präsentierte in einer weiteren Arbeit eine lincRNA mit einer Länge von 2,2 kb (2200 Nukleotide), für die eine Funktion bei der Steuerung von Chromatin-Komplexen (Material aus dem die Chromosomen bestehen, Komplexe aus DNA und Protein) innerhalb der Zelle aufgezeigt werden konnte (Rinn et al. 2007). Das war erst die vierte lincRNA, für die eine Funktion nachgewiesen war (Blow 2009).
Nun hat ein größeres Forscherteam um Rinn (Guttmann et al. 2009) mehr als 1000 lincRNAs in verschiedenen Zellen von Säugetieren (Mäusen) nachgewiesen. Diese lincRNAs liegen eingestreut zwischen Protein-codierenden Bereichen und werden abhängig vom Zustand des Chromatins (d.h. vom Zelltyp) in RNA umgeschrieben. Die Autoren entwickelten neue Methoden zur Funktionsanalyse, mit deren Hilfe sie für ca. 150 lincRNAs deren Beteiligung an verschiedensten Prozessen von der Variabilität (Pluripotenz) embryonaler Stammzellen bis hin zur Zellvermehrung nachweisen konnten (für 85 davon konnte dies in unabhängigen Experimenten bestätigt werden). Für einige dieser nicht-codierenden RNAs konnte gezeigt werden, dass deren Transkription durch spezielle Faktoren (z. B. p53, Sox2, Oct4) reguliert wird. Vor allem betonen Guttmann et al., dass diese Sequenzbereiche hinsichtlich ihrer Anordnung im Genom (genomic loci), ihrer in RNA umgeschriebenen Sequenz (exonic sequence) und den zugehörigen Steuer- und Regulationsbereichen (promotor regions) unter hohem Selektionsdruck zu stehen scheinen. Das bedeutet: sie sind in ihrer Sequenz zu mehr als 95 % konserviert, sie zeigen beim Vergleich zwischen 21 Genomen von Säugetieren nur sehr geringe Substitution*. Die auffällig geringe Variation in der Sequenz wird hier also als starker Hinweis auf biologische Aktivität gewertet.
*Substitution (= Änderung einzelner Bausteine)
Die Autoren spekulieren insgesamt über eine Funktion der lincRNAs als Transkriptionsfaktoren, dass also diese RNA-Moleküle regulierende Aufgaben in der Umschreibung von DNA-Abschnitten ausüben. Dafür gibt es bisher einige Hinweise, die allerdings weiterer experimenteller Überprüfungen und Bestätigungen bedürfen.
Mit den von Guttmann et al. (2009) vorgelegten Befunden und deren Interpretation wird die Aufgabe noch dringlicher, über neue, bisher nicht etablierte Aufgaben von Genomabschnitten nachzudenken und Methoden zu deren Aufklärung zu entwickeln. Die wertende Bezeichnung als „junk DNA“ hat sich als voreilig (und nicht innovativ) erwiesen. Auch zukünftig – so steht zu erwarten – kann man mit Phantasie und Kreativität in Verbindung mit sorgfältigen Untersuchungsmethoden noch manche bisher unbekannten Funktionen im Genom finden.
Literatur
Blow N (2009) Rethinking junk DNA. Nature 458, 240-241.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein B, Kellis M, Regev A, Rinn JL, Lander ES (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223-227.
Rinn JL, Euskirchen G, Bertone P, Martone R, Luscombe NM, Hartman S, Harrison PM, Nelson FK, Miller P, Gerstein M, Weissmann S, Snyder M (2003) The transcriptional activity of human Chromosome 22. Genes Dev. 17, 529-540.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX Loci by noncodimg RNAs. Cell 129, 1311-1323.
Autor dieserNews: Harald Binder
© 2009, http://www.genesisnet.info/schoepfung_evolution/n125.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
07.10.08 C4-Evolution: Makroevolution oder Aktivierung vorhandener Gene?
In Heft 1 des Jahrgangs 2008 der Zeitschrift Studium Integrale Journal (Stud. Int. J. 15, 3-17) erschien zu dieser Problematik ein ausführlicher Artikel von Dr. Herfried Kutzelnigg unter der Überschrift „Zur Evolution der C4-Pflanzen. Ist C4-Photosynthese 45-mal unabhängig voneinander entstanden?“. Wir wollen hier einige allgemein interessierende Aspekte dieses Themas vorstellen. Details zur Biochemie, Ökophysiologie, Anatomie, Genetik und Evolution sowie zahlreiche Abbildungen und Literaturhinweise sind dem genannten Beitrag zu entnehmen.
Allgemein bekannt ist der Standardfall der Photosynthese, der auch als C3-Photosynthese bezeichnet wird, weil die ersten fassbaren Reaktionsprodukte C3-Körper sind, d.h. organische Verbindungen mit 3 C-Atomen. Weniger bekannt ist, dass es neben diesem Standardtyp noch zwei weitere Typen gibt, nämlich die C4-Photosynthese und die CAM-Photosynthese. Erstere ist danach benannt, dass das erste fassbare Reaktionsprodukt nicht wie bei der typischen Photosynthese ein Molekül mit 3 C-Atomen, sondern eines mit 4 C-Atomen ist, letztere danach, dass sie zuerst bei Dickblattgewächsen (Crassulaceae) entdeckt wurde und mit deren auffälligen Säurestoffwechsel (Acid Metabolism) in Zusammenhang steht, bei dem nachts Säuren angereichert werden. Beiden Photosynthesewegen ist gemeinsam, dass der eigentlichen Photosynthese ein Prozess vorgeschaltet ist, der zunächst Kohlendioxid speichert, um es später in angereicherter Form wieder dem von der Standardphotosynthese bekannten Stoffaufbau zur Verfügung zu stellen. Letzterer Teilschritt ist bei den C4-Pflanzen räumlich und bei den CAM-Pflanzen zeitlich getrennt. Dem C4-Weg folgen knapp 8.000 Arten bzw. 2-3 % aller Blütenpflanzen, und bei der CAM-Photosynthese sind es sogar 16.000 bis 20.000 Arten.
Im Folgenden wollen wir uns auf die C4-Pflanzen beschränken. Sie stellen eine Anpassung an Standorte mit hohen Temperaturen und hoher Lichtintensität dar und sind dort der C3-Photosynthese gegenüber deutlich überlegen. Bekannte Beispiele sind Mais, Zuckerrohr und Hirse-Arten. Auffälliges Merkmal ist im typischen Fall die starke Vergrößerung jener Zellen, die die Leitbündel umgeben (Bündelscheidenzellen) und wegen ihrer kranzartigen Anordnung auch Kranzzellen genannt werden. Ihre Chloroplasten sind meist sehr groß und haben nicht die sonst üblichen Grana (= Membranstapel).
Man hat festgestellt, dass alle entscheidenden Enzyme der C4-Photosynthese grundsätzlich auch bei C3-Pflanzen vorkommen, allerdings in abgewandelter Form als so genannte Isoenzyme, und dass diese in anderen Zellorganellen ihre Funktion ausüben, als man es sonst kennt. Die C4-typische PEP-Carboxylase z. B. findet sich in geringer Menge auch bei C3-Pflanzen, wo ihr aber ganz andere Aufgaben zukommen als bei C4-Pflanzen.
C4-Arten sind breit verstreut und unsystematisch über das System der Blütenpflanzen verteilt. Würde man ihr Vorkommen in einen der „Stammbäume“ der Blütenpflanzen eintragen, käme dabei heraus, dass diese komplexe Erscheinung mindestens 45-mal in der Evolution unabhängig voneinander entwickelt worden sein müsste. Bedenkt man, wie gering die Wahrscheinlichkeit ist, dass ein komplexes System durch ungerichtete Mutationen neu entsteht (vgl. „Mutation“, https://www.genesisnet.info/index.php?Artikel=41241&Sprache=de&l=1), und um wie viel unwahrscheinlicher es ist, dass solche Systeme gleich mehrmals unabhängig voneinander (konvergent) entstehen, ist eine 45-malige Neubildung eine echte Herausforderung an evolutionäre Mechanismen. Das ist Biologen auch schon lange aufgefallen. Da keine grundsätzlich neuen Enzyme erforderlich sind, wird heute gerne argumentiert, dass es sich nur um kleinere Veränderungen handeln dürfte, namentlich solche im Bereich der Regulation (vgl. „Homeobox-Gene und Evolution“, https://www.genesisnet.info/index.php?Artikel=41265&Sprache=de&l=1). Wie dem auch sei, muss doch bedacht werden, dass die Neueinführung des C4-Zusatzweges mit seinen zahlreichen neuen Strukturen und Einsatzorten und neuen Enzymformen doch eines ausgeklügelten Gesamtprogramms bedarf, das außerdem garantieren muss, dass sich eventuelle Zwischenstufen nicht gegenseitig behindern. Dennoch hält die Mehrzahl derer, die sich dazu äußern, an einer unabhängigen de novo-Entstehung fest. Es gibt aber auch Stimmen, die sich dafür aussprechen, dass die zugrunde liegenden genetischen Gegebenheiten schon vor dem Sichtbarwerden des Phänomens vorhanden waren. Man spricht dann von „uralten Genen“, die also schon bei den gemeinsamen Vorfahren existierten, aber dort nicht realisiert wurden.
Vorstellungen dieser Art erhalten starken Auftrieb durch neuere Forschungen, bei denen vollständige Genome von Organismen sequenziert wurden, und bei denen sich immer wieder herausstellte, dass auch einfacher gebaute Lebewesen oft viel mehr an genetischer Information besitzen, als sie aktuell realisieren. Als Beispiel sei auf eine Seeigel-Art hingewiesen, bei der 3 % des Genoms für die Photorezeption zuständig sind, obwohl Seeigel gar keine Lichtsinnesorgane besitzen und auch nur begrenzt auf Licht reagieren.
Im Rahmen der Grundtypenbiologie („Heutige Grundtypen“, https://www.genesisnet.info/index.php?Artikel=1241&Sprache=de&l=1) spricht man von „latenten“ Genen, da der Begriff „alte Gene“ einen Abstammungszusammenhang voraussetzt, der aber nicht bewiesen ist. Von der Sache her handelt es sich aber um dasselbe, und da diese Vorstellung gut auch im Rahmen der Evolutionstheorie gedeutet werden kann, ist davon auszugehen, dass die Existenz vorvorhandener Gene als Zeichen eines gemeinsamen genetischen Erbes bald auch allgemein akzeptiert sein wird, und dass bald die Stimmen derer leiser werden, die jedem Sichtbarwerden eines Bauplans auf eine evolutive Neuentstehung zurückführen wollen.
In der Schöpfungsforschung wurde als Hypothese formuliert, dass die Ausgangspopulationen der Grundtypen genetisch polyvalent waren, also ausgerüstet mit vielfältigen Möglichkeiten ihres Erbgutes („Genetisch polyvalente Stammformen von Grundtypen“, https://www.genesisnet.info/index.php?Artikel=1244&Sprache=de&l=1). Diese Annahme hat sich immer wieder bestätigt. Latente Gene gehören in diesen Bereich. Sie sind quasi als Reserve bereits in irgendeiner Form vorhanden und können bei Bedarf aktiviert werden. Das ist so ähnlich, wie der Hochgebirgstourist für den Fall eines Falles manches in seinen Rucksack hineinsteckt, was er aber in der Regel unbenutzt wieder zurückträgt.
Dass Organismen einen bestimmten Vorrat an Genen haben, die sie im Augenblick nicht benötigen, aber bei entsprechenden Gelegenheiten aktivieren, erweist sich bei näherem Hinsehen als weit verbreitet, so dass die Idee des parallelen Vorkommens der Anlagen für den C4-Weg neben denen für den C3-Weg nicht als ungewöhnlich anzusehen ist. Einige Beispiele aus dem Bereich der Blütenpflanzen mögen das illustrieren:
- Der Wasser-Hahnenfuß bildet unter Wasser völlig andere Blätter aus als auf der Wasseroberfläche oder in der Luft.
- Der Wasser-Knöterich kann in einer Landform auf Äckern wachsen oder als Schwimmform im Wasser und sieht dann völlig verschieden aus.
- Gallwespen können in Blättern von Pflanzen die Bildung von Organen, sogenannten Gallen induzieren, die so bei keiner Pflanze beobachtet werden. Die Möglichkeit zur Ausbildung der spezifischen Gallbildung muss im Erbgut der Pflanze verborgen vorhanden sein.
- Innerhalb von Gattungen findet man nicht selten sowohl krautige als auch holzige Vertreter mit ihren sehr unterschiedlichen genetischen Ausrüstungen, und zwar breit gestreut über das System der Zweikeimblättrigen Blütenpflanzen.
Das weit verstreute Vorkommen der C4-Photosynthese (und das gilt mindestens genauso für die CAM-Photosynthese) lässt sich also recht gut durch die Annahme latent vorhandener Gene erklären. Ob diese hypothetische Annahme richtig ist oder nicht oder eventuell modifiziert werden müsste, könnte in der Zukunft im Rahmen genetischer Analysen eindeutig geklärt werden, zumal in dieser Richtung zurzeit intensiv geforscht wird. Solchen Ergebnissen ist daher mit Spannung entgegenzusehen.
Trotz der geschilderten Unwahrscheinlichkeit der wiederholten Neubildung des C4-Komplexes wird diese hin und wieder als Beispiel für Makroevolution und damit auch ausdrücklich als Argument gegen Schöpfung herangezogen. Man argumentiert dabei, dass der für Makroevolution notwendige Selektionsdruck deshalb so extrem hoch sei, weil C3-Photosynthese mit ihrem Schlüsselenzym Rubisco (Ribulose-1.5.bisphospat-Carboxylase-Oxygenase) eine Fehlkonstruktion sei, und sich deren Ersatz durch C4-Photosynthese mit ihrem Schlüsselenzym PEP-Carboxylase daher schnell durchsetzen kann. Gegen die Abstempelung der Rubisco als Fehlkonstruktion gibt es allerdings zahlreiche starke Argumente, die in der eingangs zitierten ausführlichen Darstellung nachgelesen werden können. An dieser Stelle sei nur darauf hingewiesen, dass ein Selektionsvorteil nur unter sehr speziellen klimatischen Bedingungen gegeben ist, und dass die Idee der Fehlkonstruktion der Rubisco u.a. auch durch enzymkinetische Studien widerlegt wurde. Außerdem darf nicht übersehen werden, dass auch alle C4-Pflanzen für die eigentliche Photosynthese auf die Rubisco angewiesen sind.
Zusammenfassung (übernommen aus der ausführlichen Version in Studium Integrale Journal):
Neben der gewöhnlichen Photosynthese (C3-Photosynthese) gibt es als weiteren Typ noch die so genannte C4-Photosynthese, die sich biochemisch und anatomisch deutlich unterscheidet. Diesem zweiten Weg folgen etwa 2-3% aller Blütenpflanzen. Sie sind breit über das System verteilt, so dass heute eine mindestens 45-malige Neuentstehung angenommen wird.
C4-Photosynthese ist bei hohen Temperaturen und hoher Lichtintensität der C3-Photosynthese deutlich überlegen. Es ist aber nicht so, wie verschiedentlich behauptet wird, dass C4-Photosynthese der evolutiv fortgeschrittene Weg ist, während C3-Photosynthese und das mit ihr assoziierte Enzym Rubisco eine Fehlkonstruktion darstellen. Die gängige Vorstellung über die Entstehung der C4-Photosynthese ist die, dass die zahlreichen dafür notwendigen genetischen Voraussetzungen immer wieder konvergent neu entstanden sind. Dies ist aber in hohem Maße unwahrscheinlich.
Der vorliegende Artikel bespricht die zum Verständnis der Herkunft des C4-Weges nötigen Grundlagen der Anatomie, Biochemie, Ökophysiologie und Entwicklungsbiologie und schlägt als alternatives Erklärungsmodell für die Entstehung der C4-Photosynthese vor, dass der C4-Komplex jeweils im Erbgut der Vorfahren bereits latent vorhanden war, um im Bedarfsfall aktiviert zu werden.
Die neueste Ausgabe von Studium Integrale Journal mit dem ausführlichen Artikel über C4-Photosyntehse ist bei der SG Wort und Wissen erhältlich (http://www.wort-und-wissen.de/sij).
Autor dieserNews: Studiengemeinschaft Wort und Wissen
© 2008, http://www.genesisnet.info/schoepfung_evolution/n119.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
19.05.08 Das Erbgut des Schnabeltiers: Gen-Mosaik?
Täglich wird die Liste der Organismen, deren Genom (= Erbgut) sequenziert wurde, erweitert, so dass die Nachricht über ein neu analysiertes Genom meistens keine Aufmerksamkeit mehr erweckt und im Rauschen der vielen Daten untergeht. Dennoch fand das jüngst veröffentlichte Genom des Schnabeltiers (Ornithorhynchus anatinus) von vielen populären Wissenschaftsmedien starke Beachtung und interessante Details wurden einer breiten Öffentlichkeit zugänglich gemacht. Die große Aufmerksamkeit liegt vor allem in der Besonderheit dieses Lebewesens begründet. Das Schnabeltier (engl. platypus) wird den Säugetieren zugeordnet, weil die weiblichen Tiere Milch produzieren und der Nachwuchs über Hautflecken mit Drüsenfunktion gestillt werden kann. Darüber hinaus zeigt es eine einzigartige Kombination von Merkmalen: sein Pelzkleid unterstreicht die Säugernatur, die Tatsache, dass die jungen Schnabeltiere aus Eiern schlüpfen, rückt es eher in die Nähe von Reptilien oder Vögel. Auch seine Nase, die in einer an einen Entenschnabel erinnernden Form ausgebildet ist, legt einen Vergleich mit Vögeln nahe. Dieser Schnabel enthält ein komplexes System von Sensoren für mechanische und elektrische Signale, die es dem Tier erlauben, im Wasser mit geschlossenen Augen seine Nahrung zu erbeuten, die aus Insektenlarven, Würmern und Krabben besteht. Der Schwanz des Tieres ähnelt dem eines Bibers. An den Hinterbeinen findet sich bei erwachsenen Männchen ein Sporn, in dem das Tier Gift für Auseinandersetzungen bereithält, ein sehr seltenes Kennzeichen bei Säugern, eher typisch für Reptilien. Die Bezeichnung als Kloakentier (Monotremata) beruht darauf, dass bei diesen Tieren – wie bei den Vögeln und vielen Reptilien – nur eine Öffnung für die Geschlechtsorgane, den Harnleiter und den Darm vorhanden ist.
Beim Schnabeltier sind also Merkmale kombiniert, die man typischerweise von ganz verschiedenen Tierklassen kennt. Diese mosaikartige1 Zusammensetzung macht das Schnabeltier zu einer Herausforderung für Biologen, die sich mit der Systematik von Organismen beschäftigen. Auf diesen Sachverhalt weisen die Autoren der Studie über das Genom des Schnabeltiers am Eingang ihrer Veröffentlichung noch einmal ausdrücklich hin und betonen damit die mit dem Genom verknüpften Hoffnungen auf neue Erkenntnisse.
Das Genom des Schnabeltiers liegt verteilt auf 52 Chromosomen vor, wobei es sich um wenige Makro- und viele Mikrochromosomen handelt, darunter 10 Geschlechtschromosomen.
Anzahl der Gene und Geruchssinn. Aufgrund der bisherigen Untersuchungen enthält das Genom 18.527 proteinkodierende Gene, d.h. bei einem Genom aus ca. 2,1 x 109 Nukleotiden (ca. 2/3 der Größe des menschlichen Genoms von ca. 3.1 x 109 Nukleotiden) enthält das Genom des Schnabeltiers größenordnungsmäßig etwa dieselbe Anzahl von Genen wie der Mensch. Überraschend war die hohe Zahl der kodierten Chemorezeptoren und damit der unerwartet gut ausgebildete Geruchssinn, den man bei einem Tier nicht erwartet, das die meiste Zeit im Wasser verbringt. In dieser Hinsicht erweist sich das Schnabeltier den Säugern ähnlicher als den Sauropsidae (eine systematische Gruppe der Landwirbeltiere, die Reptilien und Vögel – sowie deren fossile Vertreter – umfasst).
Eier. Die Eier des Schnabeltiers sind im Vergleich zu denjenigen von Reptilien und Vögeln klein (Durchmesser ca. 4 mm), sie sind wie bei allen Säugern von einer Glashaut (Zona Pellucida) umhüllt. Im Genom des Schnabeltiers sind alle vier vom Menschen bekannten Gene der Glashautproteine kodiert. Die Zahl der kodierten Eidotter-Proteine (Vitellogenine) ist verglichen mit Sauropsidae reduziert; das Ei erscheint damit nicht für eine längerfristige Nahrungsgrundlage des darin befindlichen Embryos geeignet.
Zeitgleich mit der Veröffentlichung in Nature wurden in der Zeitschrift Genome Research mehrere Fachpublikationen zu speziellen Aspekten des Schnabeltier-Genoms (als online-Version) publiziert (Murchison et al. 2008; Park et al. 2008; Schmitz et al. 2008; Veyrunes et al. 2008; Whittington et al. 2008).
mikro-RNA. Murchison et al. 2008 und Schmitz et al. 2008 konzentrierten sich auf die Genabschnitte, in denen kleine RNA-Abschnitte kodiert sind, sogenannte mikro-RNA, die für die Regulation sehr bedeutsam sind. Muchison et al. (2008) merken in ihrer Arbeit an: „Wir fanden, dass im Schnabeltier mikro-RNA, die es nur mit anderen Säugern gemeinsam hat, aber auch solche, die es nur mit Vögeln und Reptilien teilt.“2 und „Damit zeigen sich die ungewöhnlichen morphologischen Eigenschaften auch auf der Ebene des Genoms und sogar auf dem Niveau kleiner RNA-Fragmente.“
Milch. Die Zusammensetzung der Milch des säugenden Schnabeltiers ist ähnlich komplex wie diejenige von Säugern, d.h. sie enthält Zucker, Fette, Milchproteine zur Ernährung und mit antimikrobiellen und weiteren bioaktiven Funktionen. Die Gene für Milcheiweiß-Proteine (Casein) sind im Genom ähnlich wie bei anderen Säugern zusammengruppiert. Bei Säugern stellt man sich vor, dass die Caseingene aus Genen für Zahnschmelzproteine (Enameline) durch Genduplikation hervorgegangen sind. Während adulten (= ausgewachsenen) Schnabeltieren Zähne fehlen, sind diese bei Jungtieren sowie bei fossilen Formen vorhanden.
Gift. Giftproduktion ist bei Säugetieren nur in ganz wenigen Ausnahmefällen beschrieben. Erwachsene Schnabeltier-Männchen können Gift nicht durch einen Biss, sondern mit dem erwähnten Sporn am Hinterbein zum Einsatz bringen. Bisher ist bekannt, dass das Gift des Schnabeltiers einen Cocktail aus mindestens 19 verschiedenen Substanzen darstellt. Die Autoren interpretieren die zugrunde liegenden genetischen Daten als Beispiel für konvergente (d. h. unabhängige) Evolution von Giften in Reptilien und Kloakentieren. Die Gene für die Giftkomponente scheinen durch Genduplikation aus antimikrobiell wirksamen ß-Defensinen hervorgegangen zu sein. Auch Giftstoffe von Schlangen sollen laut K. Belov, einer der Autorinnen von Whittington et al. 2008, durch andere Genduplikationen entstanden sein. Für Belov stellt dies ein „überwältigendes Beispiel für konvergente Evolution“3 dar. Obwohl damit überhaupt nicht verstanden ist, warum das Schnabeltier als eines der auffällig wenigen Beispiele unter den Säugetieren überhaupt Giftstoffe bildet.
Genetische Hinweise auf das Immunsystem zeigen die erwartete Ähnlichkeit zwischen Schnabeltier und anderen Säugern, wobei im Genom des Schnabeltiers mit 214 natürlichen Killerzellenrezeptoren viel mehr Gene vorliegen als beim Menschen (15) oder der Ratte (45).
Chromosomen. Die Chromosomen des Schnabeltiers legen eine Beziehung sowohl zu Säugetieren als auch zu Reptilien nahe. Bei den 52 Chromosomen des Schnabeltiers brachte die Analyse keine Hinweise auf eine Korrelation zwischen der Position von orthologe Gene4 auf den kleinen Schnabeltier-Chromosomen und den Mikrochromosomen von Hühnchen. Die 5 X-Chromosomen zeigen auffällige Ähnlichkeit mit dem Z-Chromosom von Hühnchen und eine geringe im Vergleich zum menschlichen X-Chromosom, was die Autoren als Hinweis dafür interpretieren, dass das X-Chromosom des Schnabeltiers auf das eines vogelähnlichen Reptilienvorläufers zurückgeht.
Schlussfolgerungen. Warren et al. (2008) schreiben in ihren abschließenden Schlussfolgerungen: „Seit der erstmaligen Beschreibung des Schnabeltiers fällt es auf als eine Art mit vermischten Eigenschaften aus Reptilien und Säugetieren, diese Besonderheit findet sich auch auf der Ebene des Genoms wieder.“5
Angesichts dieser Feststellung, dass sich die mosaikartig zusammengesetzten Charakteristika aus Reptilien- und Säuger-typischen Elementen beim Schnabeltier auch auf der genetischen Ebene zeigen, fällt es nicht leicht, die optimistische Erwartung der Autoren zu teilen, dass das Genom des Schnabeltiers die dringend benötigten Informationen liefert, die schnellen Fortschritt bei der Untersuchung der Biologie und Evolution der Säugetiere ermöglichen sollen. Mit anderen Worten, die bisher bekannten Daten zeigen auf genetischer Ebene dieselbe außergewöhnliche Mischung von Elementen, die eigentlich für typisch für Reptilien, Vögel oder Säugetiere sind. Auf genetischer Ebene spiegelt sich also – nicht ganz unerwartet – das wieder, was aus der bisherigen morphologischen (= gestaltlichen) und anatomischen Beschreibung des Schnabeltiers bereits bekannt war: eine mosaikartige Zusammensetzung von Merkmalen, die jedes für sich in anderen Organismen oder ganzen Organismengruppen bekannt sind, aber hier in einzigartiger Weise bei einem besonderen und nur schwer einzuordnenden Lebewesen kombiniert sind. Mit dieser beschreibenden Feststellung ist allerdings entgegen häufig formulierten Andeutungen nicht etwa eine Übergangsform zwischen Reptilien und Säugetieren (und/oder Vögeln und Säugetieren?) nachgewiesen und keine Einsicht in evolutionäre Etappen im Verlauf vermuteter Entwicklungen oder gar in zugrunde liegende Mechanismen gewonnen.
Vollmundige Äußerungen über den „unbezahlbaren“ Beitrag dieser Genom-Sequenz für das Verständnis von biologischen Prozessen der Säugetierevolution (F. Collins) oder: „Das Schnabeltier-Genom hilft uns, Lücken im Verständnis zur Entwicklung des Menschen zu schließen, wie die Evolution des Y-Chromosoms“ (J. Graves) scheinen daher durch die bisher vorliegenden Daten nicht gedeckt zu sein. Vielmehr liegt der Verdacht nahe, dass Wissensfortschritte kurzerhand ohne ausreichende Begründung auch als Fortschritte im Verständnis der Evolution ausgegeben werden – vor allem von der Wissenschaftspresse.
Somit bleibt einmal mehr die Erwartung, dass noch weitere Daten notwendig sind, um die Lücken unseres Verständnisses über die Beziehungen der verschiedenen Klassen von Organismen zu verkleinern und die Einsichten in deren molekularbiologische Zusammenhänge zu erhellen. Die Behauptung „Das Genom des Schnabeltiers löst die Geheimnisse der Evolution der Säugetiere“6 (Genome Research) ist derzeit jedenfalls noch nicht eingelöst.
Anmerkungen
1 Warren et al. (2008) gebrauchen den Begriff „Amalgam“ (Quecksilber-Legierungen): „The platypus genome, as well as the animal, is an amalgam of ancestral reptilian and derived mammalian characteristics.“
2 „Remarkably, we found that the platypus shares microRNA families uniquely with other mammals, but also uniquely with a representative of birds and reptiles,“ und „Thus, the unusual morphology of these animals is also reflected at the genomic level and at the level of its small RNAs.“
3 „Snake venom crotamines have also evolved from beta-defensins in separate gene duplication events, making this a compelling example of convergent evolution.“
4 Orthologe Gene: meist werden darunter Gene aus verschiedenen Organismen verstanden, die funktional ähnlich sind und hohe Übereinstimmung in der Basensequenz aufweisen; sie werden als Erbstück eines gemeinsamen Vorfahren betrachtet.
5 „Since its initial description, the platypus has stood out as a species with a blend of reptilian and mammalian features, which is a characteristic that penetrates to the level of the genome sequence.“
6 „Platypus genome unravels the mysteries of mammalian evoluton
Literatur
Murchison EP, Kheradpour P, Sachidanandam R, Smith C, Hodges E, Xuan Z, Kellis M, Grützner F, Stark A, and Hannon GJ (2008) Conservation of small RNA pathways in platypus. Genome Res. doi:10.1101/gr.73056.107.
Park, J, Semyonov J, Chang CL, Yi W, Warren W, and Hsu SYT (2008) Origin of INSL3-mediated testicular descent in therian mammals. Genome Res. doi:10.1101/gr.7119108.
Schmitz, J, Zemann A, Churakov G, Kuhl H, Grützner F, Reinhardt R, and Brosius J (2008) Retroposed SNOfall – A mammalian-wide comparison of platypus snoRNAs. Genome Res. doi:10.1101/gr.7177908.
Veyrunes F, Waters PD, Miethke P, Rens W, McMillan D, Alsop AE, Grützner F, Deakin JE, Whittington CM, Schatzkamer K, Kremitzki CL, Graves T, Ferguson-Smith MA, Warren W, Graves JAM (2008) Bird-like sex chromosomes of platypus imply recent origin of mammal sex chromosomes. Genome Res. doi:10.1101/gr.7101908.
Warren WC et. al (2008) Genome analysis of the platypus reveals unique signatures of evolution. Nature 453, 175- 184. Umfangreiche Supplementary Information online unter: http://www.nature.com/nature/journal/v453/n7192/suppinfo/nature06936.html
Whittington CM, Papenfuss AT, Bansal P, Torres, AM, Wong ESW, Deakin JE, Graves T, Alsop A, Schatzkamer K, Kremitzki C, Ponting CP, Temple-Smith P, Warren WC, Kuchel PW, and Belov, K (2008) Defenisns and the convergent evolution of platypus and reptile venom genes. Genome Res. doi:10.1101/gr.7149808
Autor dieserNews: Harald Binder
© 2008, http://www.genesisnet.info/schoepfung_evolution/n112.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
20.12.07 Genomforschung – Wieviel „Schrott“ bleibt übrig?
Oder: Wie findet man bisher übersehene bedeutsame DNA-Sequenzen?
Der erstaunlich geringe Anteil codierender DNA-Sequenzen am Genom (Erbgut) eines Organismus war für manchen Wissenschaftler in der Vergangenheit Anlass, von „Abfall“ („junk“) zu reden – Anteile des Erbguts, die im Laufe der Evolution angesammelt wurden und überflüssig geworden seien. Diese Deutung war zwar nicht unumstritten, aber doch – vor allem in populären Medien – weit verbreitet. Die Anzeichen dafür mehren sich jedoch, dass immer weitere Teile des Genoms von Bedeutung sind. Aber wie kann man analytisch DNA-Abschnitte, die nicht in Proteine übersetzt werden (Translation), überhaupt erkennen? McCallion et al. (2007) haben eine Methode entwickelt, diese im Bereich eines Gens angewendet und dabei überraschende Erfahrungen gemacht.
Die Autoren untersuchten beim Zebrafisch (Danio rerio) die Umgebung des Gens phox2b. Dieses Gen spielt u. a. bei der Entwicklung von Nervenzellen eine Rolle, bei der Stressverarbeitung und im Verdauungssystem. DNA-Fragmente (48 Amplikons) aus diesem Bereich transferierten die Forscher gekoppelt mit Gen eines grün fluoreszierenden Proteins (GFP) in Embryonen des Zebrafisches. In dem von ihnen etablierten Testsystem gibt sich ein regulatorischer DNA-Abschnitt dadurch zu erkennen, dass der Embryo GFP produziert und damit grün fluoresziert. In 17 Fällen erhielten McCallion et al. fluoreszierende Zebrafisch-Embryonen.
Mit den fünf gebräuchlichsten Programmen zur Genomanalyse konnten nur zwischen 29 und 61 % der tatsächlich gefundenen regulatorisch wirksamen DNA-Sequenzen erkannt und prognostiziert werden. Damit ist gezeigt, dass die bisher verfügbaren Computerprogramme weiterentwickelt werden und auf eine solidere experimentelle Basis gestellt werden müssen. Diese Resultate unterstreichen auch den großen Bedarf an weiteren methodischen Zugängen, um bedeutsame DNA-Bereiche zu identifizieren. Durch Anwendung der bereits verfügbaren Techniken auf weitere Genbereiche ist ein erheblicher Kenntniszuwachs über weitere funktionelle Sequenzen zu erwarten.
Sollte ein erheblicher Anteil der im Genom niedergelegten Information bisher unerkannt sein, hätte das auch weitreichende Konsequenzen für die Anwendung molekularbiologischer Daten für die Analyse molekularer Stammbäume. Dann müsste – auch darauf deuten die Autoren hin – bei der Interpretation molekularbiologischer Daten im Sinne phylogenetischer Fragestellungen im Lichte dieser Erkenntnisse sehr viel sorgfältiger vorgegangen werden.
Quelle:
McGaughey DM, Vinton RM, Huynh J, Al-Saif A, Beer MA & McCallion AS (2007) Metrics of sequence constraint overlook regulatory sequences in an exhaustive analysis at phox2b. Genome Res. (download advance articles 10.12.2007): http://www.genome.org/cgi/reprint/gr.6929408v1
Autor dieser News: Harald Binder
© 2007, http://www.genesisnet.info/schoepfung_evolution/n102.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
11.08.05 Neuer Artikel über molekulare Stammbäume und molekulare Uhren
Ein häufig genanntes Argument für Evolution ist die vermeintliche Übereinstimmung molekularer Ähnlichkeitsbäume (Dendrogramme) mit klassischen Stammbäumen, die vor allem auf dem äußeren Bau der Lebewesen beruhen. Tatsächlich ist aber diese Übereinstimmung in der Regel nur teilweise gegeben. Auch das Konzept der „molekularen Uhr“ wird in der Fachwelt kontrovers diskutiert. Im Artikel „Molekularbiologie“ (https://www.genesisnet.info/index.php?Artikel=41302&Sprache=de&l=2) der Studiengemeinschaft Wort und Wissen wird diese Thematik ausführlich dargestellt. Bisher liegt der Beitrag nur als Expertenartikel vor. Folgende Schlussfolgerungen werden gezogen:
- Die Ähnlichkeiten von Makromolekülen (Aminosäure- und DNS-Sequenzen) können zwar prinzipiell evolutionstheoretisch gedeutet werden; dies ist aber nicht zwingend.
- Merkmalsauswahl und Stammbaumrekonstruktion sind komplizierte Verfahren, deren Vorannahmen nicht zutreffen müssen und die einige subjektive Entscheidungen erfordern. Eine voraussetzungslose Konstruktion von Verwandtschaft mittels molekularer Daten ist nicht möglich.
- Widersprüche zwischen den Rekonstruktionen der Verwandtschaftsbeziehungen können vielfältige Ursachen haben: Fehler bei der Auswahl der Arten oder der Datenanalyse, rasche Artbildung, polymorphe Stammpopulationen, Hybridisierung, horizontaler Gentransfer, Konvergenzen. Welche Erklärung(en) im Einzelfall zutrifft (zutreffen), kann oft nicht eindeutig entschieden werden.
- Jede Stammbaumrekonstruktion – molekular oder durch andere Daten begründet – basiert auf der Annahme, dass die verglichenen Merkmale (Sequenzen) phylogenetisch homolog sind, d. h. dass ihre Ähnlichkeiten bzw. Gemeinsamkeiten abstammungsbedingt sind. Falls die Ähnlichkeit aber eher funktionelle Erfordernisse widerspiegelt, geben die ermittelten Bäume nur die Ähnlichkeit der Gene der sie tragenden Organismen wieder, nicht aber deren Abstammungsverwandtschaft.
- Eine globale molekulare Uhr existiert entgegen den ursprünglichen Erwartungen nicht. Lokale Uhren, die sich auf ein bestimmtes Gen und eine bestimmte Auswahl von Organismen beziehen, gibt es zwar, aber jeder Datensatz muss daraufhin untersucht werden, ob variierende Evolutionsraten vorliegen.
- Auch molekulare Uhren beruhen auf der Grundvoraussetzung von Evolution und können daher nicht als unabhängiger Beleg für Evolution gelten.
- Molekulare Systematik hat sehr erfolgreich viele Verwandtschaftsbeziehungen klären können. Die besten Ergebnisse liegen im mikroevolutiven Bereich (Mikro- und Makroevolution, https://www.genesisnet.info/index.php?Artikel=41223&Sprache=de&l=1), d.h. innerhalb vermuteter Grundtypen (Artbegriffe, https://www.genesisnet.info/index.php?Artikel=41224&Sprache=de&l=1). Die Gründe für eine schlechte Auflösung von Verwandtschaftsverhältnissen sind sehr unterschiedlich, je nachdem ob eine nahe oder entfernte Verwandtschaft vorliegt. Bei naher Verwandtschaft können Polymorphismen (= genetische Vielseitigkeit) in der Stammpopulation, rasche Artbildung, Hybridisierung (= Kreuzungen) oder horizontaler Gentransfer die Ursache sein, bei ferner Verwandtschaft Konvergenz, postulierter hypothetischer horizontaler Gentransfer oder andere unklare Gründe-
Autor dieserNews: Reinhard Junker
© 2005, http://www.genesisnet.info/schoepfung_evolution/n44.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
11.07.05 Neuer Artikel über Evolution virtueller Lebewesen
Die Süddeutsche Zeitung berichtet in ihrer Artikelserie „Streitfall Evolution“ im Beitrag „Skepsis inklusive“ am 1. 7. 2005 vom Evolutionsprogramm „Avida“. Die Autorin Tina Baier meint, dieses Programm ermögliche es, Evolution im Zeitraffer zu simulieren und zeige, dass die Evolution in der Lage sei, komplexe Strukturen zu entwickeln. „Tausende von Kreationisten haben es sich schon heruntergeladen, um einen Fehler zu finden – bisher ohne Erfolg.“ Die Frage ist jedoch nicht, ob das Programm fehlerhaft ist, sondern was es wirklich leistet. Zwei Mitarbeiter der Studiengemeinschaft Wort und Wissen haben sich mit dieser Materie anhand eines vielbeachteten Nature-Artikels von Richard E. Lenski beschäftigt. Die Ergebnisse wurden Ende Mai in der neuesten Ausgabe von „Studium Integrale Journal“ (https://www.wort-und-wissen.org/publikationen/studium-integrale-journal/) veröffentlicht. Seit kurzem ist ihr Text auch online bei Genesisnet verfügbar (Evolution virtueller Lebewesen, https://www.genesisnet.info/index.php?Artikel=41246&Sprache=de&l=1).
Die kritische Analyse von Eberhard Bertsch und Torsten Waldminghaus zeigt, dass die Simulation der Evolution virtueller Lebewesen zur Abbildung der behaupteten makroevolutiven Prozesse ungeeignet ist. Ihre Kritikpunkte lauten kurzgefasst:
- Die Belohnung von Teilergebnissen auf einem evolutiven Weg kann nicht ohne Weiteres auf lebende Systeme übertragen werden.
- Die komplizierteste Funktion, die in dem beschriebenen System entstehen kann, ist kaum mit der Komplexität eines sehr einfachen Proteins zu vergleichen.
- Die Simulation der Entstehung komplizierterer Eiweiße ist mit diesem System unmöglich.
- Eine zentrale Vorgabe der virtuellen Organismen ist den natürlichen Bedingungen entgegengesetzt.
Die Autoren kommen zum Ergebnis, dass der Ansatz von Lenski zwar insofern eine sehr gute Idee ist, als er die grundlegende Unterscheidung von Syntax und Semantik in die Ursprungs-Diskussion einbezieht, dass aber andererseits von „Komplexität“ der entstandenen Strukturen und Funktionen überhaupt keine Rede sein kann. Grundsätzlich bleibt auch hier das Problem offen, ob Makroevolution durch eine Aneinanderreihung von vielen mikroevolutiven Schritten entstehen kann. Eine Antwort auf die Frage nach der Möglichkeit von Makroevolution konnte mit dem beschriebenen Ansatz nicht gegeben werden.
Autor dieserNews: Reinhard Junker
© 2005, http://www.genesisnet.info/schoepfung_evolution/n40.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
17.02.05 Mechanismen der Makroevolution?
Zwei neue Artikel von Niko Winkler befassen sich mit Evolutionsmechanismen, die als Teillösungen des Problems „Makroevolution“ vorgeschlagen wurden. Mit Makroevolution ist die Entstehung neuer Baupläne oder Bauteile von Organismen gemeint; das ist etwas qualitativ anderes als bloße Variationsvorgänge, Anpassungen oder Spezialisierungen (vgl. Mikro- und Makroevolution, https://www.genesisnet.info/index.php?Artikel=41223&Sprache=de&l=1). Viele Biologen räumen ein, dass Makroevolution durch mikroevolutive Prozesse nicht erklärbar ist, und suchen nach besonderen Mechanismen für Makroevolution. Zwei Ansätze werden in den neuen Beiträgen erläutert und kritisch untersucht. Der erste Artikel handelt über das so genannte „Gene tinkering“ (s. Gene tinkering, https://www.genesisnet.info/index.php?Artikel=41264&Sprache=de&l=1). „Gene tinkering“ bedeutet „Flickschusterei mit Genen“. Einbau von Genen in andere, bereits bestehende genetische Zusammenhänge soll eine Erklärung für manche makroevolutive Veränderungen liefern. Auf diese Weise könnten genetische Netzwerke entstanden sein. Solche Vorgänge sind bislang jedoch weitgehend hypothetisch und sie erklären nicht die Herkunft der Gene an sich. Interessant ist in diesem Zusammenhang der Befund, dass biologische Netzwerke solchen Netzwerken gleichen, die durch Ingenieure erdacht wurden – ein Befund, der ein undurchdachtes gene tinkering in Frage stellt.
Im zweiten Artikel wird die Bedeutung der Homeobox-Gene für Evolution diskutiert (Homeobox-Gene und Evolution, https://www.genesisnet.info/index.php?Artikel=41265&Sprache=de&l=1). Homeobox-Gene haben eine regulative Funktion und stehen demnach am Anfang von Entwicklungsvorgängen in der individuellen Entwicklung (Ontogenese). Aufgrund dieser besonderen Funktion wurde vermutet, dass Homeobox-Gene einen Schlüssel für Makroevolution liefern könnten, weil ihr Ein- und Ausschalten erhebliche Auswirkungen auf die Gestalt hat. Doch erklären solche Vorgänge nicht, wie die von den Homeobox-Genen regulierten Gene bzw. Strukturen und die Homeobox-Gene selber ursprünglich entstanden sind.
Autor dieserNews: Reinhard Junker
© 2005, http://www.genesisnet.info/schoepfung_evolution/n33.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/
09.03.04 Neues evolutionskritisches Video von Fritz Poppenberg erschienen
Seit kurzen gibt es einen allgemeinverständlichen Vortrag von Siegfried Scherer (Autor des Genesisnet-Artikels „Wahrscheinlichkeitsbetrachtungen-Beispiel Bakterienmotor“, https://www.genesisnet.info/index.php?Artikel=41282&Sprache=de&l=1) auf Video und auf DVD. Titel: „Was Darwin nicht wissen konnte – der Streit um die Entstehung des Lebens.“
https://www.wort-und-wissen.org/produkt-kategorie/dvd/
Der Film wurde von Fritz Poppenberg, Dreilindenfilm gedreht, der auch den bekannten Videofilm „Hat die Bibel doch recht?“ produziert hat. Prof. Dr. Siegfried Scherer kommt in seinem Vortrag zu dem Ergebnis, dass die entscheidenden Fragen auch 150 Jahre nach Darwin ungelöst sind:
• Die Entstehung des Lebens ist unbekannt.
• Der Fossilbericht bietet trotz großer Mengen an Funden kaum Funde, die als Übergangsformen interpretierbar sind.
• Die Makroevolution (Höherentwicklung) ist experimentell nicht belegt.
• Trotz gegenteiliger Behauptung gilt noch immer, was der große Forscher Louis Pasteur nachwies: „Lebendes entsteht nur aus Lebendem.“
Der Vortrag ist allgemein verständlich. Daher ist „Was Darwin nicht wissen konnte“ sehr gut für Hauskreise, Jugendkreise usw. geeignet.
Dieses und ähnliche Videos können direkt bei www.wort-und-wissen.de bestellt werden.
Autor dieserNews: Reinhard Junker
© 2004, http://www.genesisnet.info/schoepfung_evolution/n15.php
Zurück zur Artikel-Übersicht: https://www.wort-und-wissen.org/publikationen/genesisnet/